Methods and compositions for treating mitochondrial diseases or disorders and heterogeneity
阅读说明:本技术 用于治疗线粒体疾病或病症和异质性的方法和组合物 (Methods and compositions for treating mitochondrial diseases or disorders and heterogeneity ) 是由 五条理志 上大介 前田秀树 于 2019-08-13 设计创作,主要内容包括:本发明提供了用于产生线粒体替换的细胞(MirC)的方法和组合物和使用这些组合物治疗患有年龄相关疾病或综合征、线粒体疾病或病症或者另外需要线粒体替换的对象的治疗方法。还提供了产生具有线粒体疾病或病症的受体细胞的方法和组合物,以及产生或提高诱导多潜能干细胞(iPSC)的产量的方法和组合物。另外,还包括提高线粒体转移的方法和组合物。(The present invention provides methods and compositions for producing mitochondrial-replaced cells (MirC) and therapeutic methods for using these compositions to treat subjects having age-related diseases or syndromes, mitochondrial diseases or disorders, or otherwise in need of mitochondrial replacement. Also provided are methods and compositions for producing recipient cells having a mitochondrial disease or disorder, and methods and compositions for producing or increasing the yield of induced pluripotent stem cells (ipscs). Additionally, methods and compositions for improving mitochondrial transport are included.)
1. A method of producing a mitochondrial-replaced cell comprising:
(a) contacting a recipient cell with an agent that reduces the copy number of endogenous mtDNA;
(b) incubating the recipient cell with the agent for a sufficient period of time to partially reduce the endogenous mtDNA copy number in the recipient cell; and
(c) co-incubating (1) the recipient cell from step (b) in which the endogenous mtDNA has been partially reduced and (2) an exogenous mitochondrion from a healthy donor for a period of time sufficient to non-invasively transfer the exogenous mitochondrion to the recipient cell, thereby producing a mitochondrion-replaced cell.
2. A method of treating a subject in need of mitochondrial replacement comprising:
(a) producing ex vivo or in vitro a mitochondrial replaced cell comprising the steps of:
(i) contacting a recipient cell with an agent that reduces mtDNA copy number;
(ii) incubating the recipient cell with the agent for a period of time sufficient to partially reduce mtDNA copy number in the recipient cell; and
(iii) (iii) co-incubating (1) the recipient cell from step (ii) in which endogenous mtDNA has been partially reduced and (2) exogenous mitochondria from a healthy donor for a period of time sufficient to non-invasively transfer exogenous mitochondria to the recipient cell, thereby producing a mitochondria-replaced cell; and
(b) administering a therapeutically effective amount of the receptor cells for mitochondrial replacement from step (a) to a subject in need of mitochondrial replacement.
3. A method of treating a subject having or suspected of having an age-related disease, the method comprising:
(a) producing ex vivo or in vitro a mitochondrial replaced cell comprising the steps of:
(i) contacting a recipient cell with an agent that reduces mtDNA copy number;
(ii) incubating the recipient cell with the agent for a period of time sufficient to partially reduce mtDNA copy number in the recipient cell; and
(iii) (iii) co-incubating (1) the recipient cell from step (ii) in which endogenous mtDNA has been partially reduced and (2) exogenous mitochondria from a healthy donor for a period of time sufficient to non-invasively transfer exogenous mitochondria to the recipient cell, thereby producing a mitochondria-replaced cell; and
(b) administering a therapeutically effective amount of the mitochondrially replaced recipient cell from step (a) to a subject having or suspected of having an age-related disorder.
4. A method of treating a subject having or suspected of having a mitochondrial disease or disorder, the method comprising:
(a) ex vivo or in vitro production of a mitochondrially replaced recipient cell comprising the steps of:
(i) contacting a recipient cell with an agent that reduces mtDNA copy number;
(ii) incubating the recipient cell with the agent for a period of time sufficient to partially reduce mtDNA copy number in the recipient cell; and
(iii) (iii) co-incubating (1) the recipient cell from step (ii) in which endogenous mtDNA has been partially reduced and (2) exogenous mitochondria from a healthy donor for a period of time sufficient to non-invasively transfer exogenous mitochondria to the recipient cell, thereby producing a mitochondria-replaced cell; and
(b) administering a therapeutically effective amount of the mitochondrial replaced recipient cell from step (a) to a subject having or suspected of having a mitochondrial disease or disorder.
5. The method of any one of claims 1-4, wherein the exogenous mitochondria are functional mitochondria.
6. The method of any one of claims 1-5, wherein the exogenous mitochondria comprise wild-type mtDNA.
7. The method of any one of claims 1-6, wherein the exogenous mitochondria are isolated mitochondria.
8. The method of claim 7, wherein the isolated mitochondria are intact mitochondria.
9. The method of any one of claims 1-8, wherein the exogenous mitochondria are allogeneic.
10. A method of producing a mitochondrial-replaced cell comprising:
(a) contacting a recipient cell with an agent that reduces the copy number of endogenous mtDNA;
(b) incubating the recipient cell with the agent for a sufficient period of time to partially reduce the endogenous mtDNA copy number in the recipient cell; and
(c) co-incubating (1) the recipient cell from step (b) in which endogenous mtDNA has been partially reduced and (2) exogenous mtDNA from a healthy donor for a period of time sufficient to non-invasively transfer exogenous mtDNA to the recipient cell, thereby producing a mitochondria-replaced cell.
11. A method of treating a subject in need of mitochondrial replacement comprising:
(a) Producing ex vivo or in vitro a mitochondrial replaced cell comprising the steps of:
(i) contacting a recipient cell with an agent that reduces mtDNA copy number;
(ii) incubating the recipient cell with the agent for a period of time sufficient to partially reduce mtDNA copy number in the recipient cell; and
(iii) (iii) co-incubating (1) the recipient cell from step (ii) in which endogenous mtDNA has been partially reduced and (2) exogenous mtDNA from a healthy donor for a period of time sufficient to non-invasively transfer exogenous mtDNA to the recipient cell, thereby producing a mitochondria-replaced cell; and
(b) administering a therapeutically effective amount of the receptor cells for mitochondrial replacement from step (a) to a subject in need of mitochondrial replacement.
12. A method of treating a subject having or suspected of having an age-related disease, the method comprising:
(a) producing ex vivo or in vitro a mitochondrial replaced cell comprising the steps of:
(i) contacting a recipient cell with an agent that reduces mtDNA copy number;
(ii) incubating the recipient cell with the agent for a period of time sufficient to partially reduce mtDNA copy number in the recipient cell; and
(iii) (iii) co-incubating (1) the recipient cell from step (ii) in which endogenous mtDNA has been partially reduced and (2) exogenous mtDNA from a healthy donor for a period of time sufficient to non-invasively transfer exogenous mtDNA to the recipient cell, thereby producing a mitochondria-replaced cell; and
(b) Administering a therapeutically effective amount of the mitochondrially replaced recipient cell from step (a) to a subject having or suspected of having an age-related disorder.
13. A method of treating a subject having or suspected of having a mitochondrial disease or disorder, the method comprising:
(a) ex vivo or in vitro production of a mitochondrially replaced recipient cell comprising the steps of:
(i) contacting a recipient cell with an agent that reduces mtDNA copy number;
(ii) incubating the recipient cell with the agent for a period of time sufficient to partially reduce mtDNA copy number in the recipient cell; and
(iii) (iii) co-incubating (1) the recipient cell from step (ii) in which endogenous mtDNA has been partially reduced and (2) exogenous mtDNA from a healthy donor for a period of time sufficient to non-invasively transfer exogenous mtDNA to the recipient cell, thereby producing a mitochondria-replaced cell; and
(b) administering a therapeutically effective amount of the mitochondrial replaced recipient cell from step (a) to a subject having or suspected of having a mitochondrial disease or disorder.
14. The method of any one of claims 1-13, wherein the agent that reduces the copy number of endogenous mtDNA is selected from the group consisting of a polynucleotide encoding a fusion protein comprising a mitochondrial-targeting sequence (MTS) and an endonuclease, a polynucleotide encoding an endonuclease, and a small molecule.
15. The method of claim 14, wherein the small molecule is a Nucleoside Reverse Transcriptase Inhibitor (NRTI).
16. The method of claim 14, wherein the polynucleotide consists of a messenger ribonucleic acid (mRNA) or a deoxyribonucleic acid (DNA).
17. The method of claim 14, wherein the recipient cell transiently expresses the fusion protein.
18. The method of claim 14, wherein the endonuclease is selected from the group consisting of XbaI, EcoRI, BamHI, HindIII, PstI, Cas9, Zinc Finger Nuclease (ZFN), and transcription activator-like effector nuclease (TALEN).
19. The method of any one of claims 14, 17 or 18, wherein the MTS targets a mitochondrial matrix protein.
20. The method of claim 19, wherein the mitochondrial matrix protein is selected from cytochrome c oxidase subunit IV, cytochrome c oxidase subunit VIII, and cytochrome c oxidase subunit X.
21. The method of any one of claims 1 to 20, wherein said agent that reduces endogenous mtDNA copy number reduces said endogenous mtDNA copy number by about 5% to about 99%.
22. The method of claim 21 wherein the agent that reduces endogenous mtDNA copy number reduces the endogenous mtDNA copy number by about 30% to about 70%.
23. The method of claim 21 wherein the agent that reduces endogenous mtDNA copy number reduces the endogenous mtDNA copy number by about 50% to about 95%.
24. The method of claim 21 wherein said agent that reduces endogenous mtDNA copy number reduces said endogenous mtDNA copy number by about 60% to about 90%.
25. The method of any one of claims 1-24, wherein the agent that reduces endogenous mtDNA copy number reduces mitochondrial material.
26. A method of producing a mitochondrial-replaced cell comprising:
(a) contacting the recipient cell with an agent that reduces mitochondrial function;
(b) incubating the recipient cell with the agent for a period of time sufficient to partially reduce endogenous mitochondrial function in the recipient cell; and
(c) co-incubating (1) the recipient cell from step (b) in which endogenous mitochondrial function has been partially reduced and (2) exogenous mitochondria from a healthy donor for a period of time sufficient to non-invasively transfer the exogenous mitochondria to the recipient cell, thereby producing a mitochondria-replaced cell.
27. A method of producing a mitochondrial-replaced cell comprising:
(a) contacting the recipient cell with an agent that reduces mitochondrial function;
(b) Incubating the recipient cell with the agent for a period of time sufficient to partially reduce endogenous mitochondrial function in the recipient cell; and
(c) co-incubating (1) the recipient cell from step (b) in which endogenous mitochondrial function has been partially reduced and (2) exogenous mtDNA from a healthy donor for a period of time sufficient to non-invasively transfer the exogenous mtDNA to the recipient cell, thereby producing a mitochondrial-replaced cell.
28. The method of claim 26 or 27, wherein the agent that reduces mitochondrial function transiently reduces endogenous mitochondrial function.
29. The method of claim 26 or 27, wherein the agent that reduces mitochondrial function permanently reduces endogenous mitochondrial function.
30. The method of claim 2 or 11, wherein the subject in need of mitochondrial replacement has dysfunctional mitochondria; a disease selected from the group consisting of: age-related diseases, mitochondrial diseases or disorders, neurodegenerative diseases, retinal diseases, diabetes, hearing disorders, genetic diseases; or a combination thereof.
31. The method of claim 30, wherein the neurodegenerative disease is selected from Amyotrophic Lateral Sclerosis (ALS), huntington's disease, alzheimer's disease, parkinson's disease, friedrich's ataxia, peroneal muscular atrophy, and cerebral leukosis.
32. The method of claim 30, wherein the retinal disease is selected from the group consisting of age-related macular degeneration, macular edema, and glaucoma.
33. The method of any one of claims 3, 12, or 30, wherein the age-related disease is selected from the group consisting of an autoimmune disease, a metabolic disease, a genetic disease, cancer, a neurodegenerative disease, and immunosenescence.
34. The method of claim 33, wherein the metabolic disease is diabetes.
35. The method of claim 33, wherein the neurodegenerative disease is alzheimer's disease or parkinson's disease.
36. The method of claim 30 or 33, wherein the genetic disease is selected from the group consisting of early senescence syndrome, vorner syndrome, and huntington's disease.
37. The method of any one of claims 4, 13, or 30, wherein the mitochondrial disease or disorder is caused by mitochondrial DNA abnormalities, nuclear DNA abnormalities, or both.
38. The method of claim 37, wherein the mitochondrial disease or disorder caused by mitochondrial DNA abnormalities is selected from Chronic Progressive External Ophthalmoplegia (CPEO), pearson Syndrome, cohn-saiya Syndrome (KSS), diabetes with deafness (DAD), mitochondrial diabetes, Leber's Hereditary Optic Neuropathy (LHON), LHON-plus (LHON-plus), neuropathy, ataxia and retinitis pigmentosa Syndrome (NARP), Maternally Inherited Leigh Syndrome (MILS), mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS), myoclonic epilepsy with ragged red fiber disease (MERRF), familial bilateral striatal necrosis/striatal nigrosis (FBSN), Luft disease, aminoglycoside-induced deafness (AID), and various deletions of mitochondrial DNA Syndrome.
39. The method of claim 37, wherein the mitochondrial disease or disorder caused by a nuclear DNA abnormality is selected from the group consisting of mitochondrial DNA deletion syndrome-4A, mitochondrial recessive ataxia syndrome (MIRAS), mitochondrial neuro-gastrointestinal encephalomyopathy (MNGIE), mitochondrial DNA deletion syndrome (MTDPS), DNA polymerase gamma (POLG) -associated disorders, sensory ataxia-dysregulated neuropathy with dysarthria and ophthalmoplegia (SANDO), leukoencephalopathy with brain stem and spinal cord involvement and elevated lactate (LBSL), coenzyme Q10 deficiency, Leigh syndrome, mitochondrial complex abnormalities, fumarate deficiency, alpha-ketoglutardialdehyde dehydrogenase complex (KGDHC) deficiency, succinyl-coa ligase deficiency, pyruvate dehydrogenase complex deficiency (PDHC), Pyruvate Carboxylase Deficiency (PCD), carnitine palmitoyl transferase i deficiency (cpt i), deficiency, and combinations thereof, Carnitine palmitoyltransferase ii (cpt ii) deficiency, carnitine-acyl-carnitine (CACT) deficiency, autosomal dominant-/autosomal recessive-progressive extra-ocular paralysis (ad-/ar-PEO), infantile spinocerebellar atrophy (IOSCA), Mitochondrial Myopathy (MM), Spinal Muscular Atrophy (SMA), growth arrest, aminouria, cholestasis, iron overload, early death (GRACILE), and Charcot-Marei-Tooth 2A (CMT 2A).
40. The method of any one of claims 1-39, wherein said endogenous mtDNA encodes a dysfunctional mitochondrion.
41. The method of any one of claims 1 to 40, wherein said endogenous mtDNA comprises mutant mtDNA.
42. The method of any one of claims 1-41, wherein said endogenous mtDNA comprises mtDNA associated with a mitochondrial disease or disorder.
43. The method of any one of claims 1-42, wherein said endogenous mtDNA is heterogeneous.
44. The method of any one of claims 1-43, wherein the recipient cell has dysfunctional endogenous mitochondria.
45. The method of any one of claims 1 to 39, wherein the endogenous mtDNA in the recipient cell comprises wild-type mtDNA.
46. The method of any one of claims 1 to 45, wherein the mitochondrially replaced cell has a total mtDNA copy number relative to the total mtDNA copy number of the recipient cell prior to contact with the agent that reduces endogenous mtDNA copy number of no greater than about 1.1 fold, about 1.2 fold, about 1.3 fold, about 1.4 fold, about 1.5 fold or more.
47. The method of any one of claims 1-46, wherein the recipient cell is an animal cell or a plant cell.
48. The method of claim 47, wherein the animal cell is a mammalian cell.
49. The method of claim 48, wherein the recipient cell is a somatic cell.
50. The method of claim 48, wherein the recipient cell is a bone marrow cell.
51. The method of claim 50, wherein the bone marrow cells are Hematopoietic Stem Cells (HSCs) or Mesenchymal Stem Cells (MSCs).
52. The method of any one of claims 1 to 49, wherein the recipient cell is a cancer cell.
53. The method of any one of claims 1 to 49, wherein the recipient cell is a primary cell.
54. The method of any one of claims 1-49, wherein the recipient cell is an immune cell.
55. The method of claim 54, wherein the immune cell is selected from the group consisting of a T cell, a phagocytic cell, a microglial cell, and a macrophage.
56. The method of claim 55, wherein the T cell is a CD4+ T cell.
57. The method of claim 55, wherein the T cell is a CD8+ T cell.
58. The method of claim 55, wherein the T cell is a Chimeric Antigen Receptor (CAR) T cell.
59. The method of any one of claims 1 to 58, wherein the transfer of exogenous mitochondrial and/or exogenous mtDNA is stable.
60. The method of claim 59, wherein said exogenous mtDNA alters heterogeneity in said recipient cell.
61. The method of any one of claims 1 to 60, further comprising delivering a small molecule, peptide or protein.
62. The method of any one of claims 1 to 61, further comprising contacting the recipient cell with a second active agent prior to co-incubating the recipient cell with exogenous mitochondria and/or exogenous mtDNA.
63. The method of claim 62, wherein the second active agent is selected from a macromolecule, a small molecule, or a cell therapy, and the second active agent is optionally selected from rapamycin, NR (nicotinamide ribose), bezafibrate, idebenone, mercaptoethylamine bitartrate (RP103), elamipramide (MTP131), omaviralone (omavelolone) (RTA408), KH176, Vatiquonone (Vatiquinone) (Epi743), lipoic acid, A0001 (alpha-tocopherolquinone), mitochondrial CoQ10(MitoQ), SkQ1(Visomitin), resveratrol, curcumin, a ketonic therapy, hypoxia, and an endocytic activator.
64. The method of claim 63, wherein the endocytic activator is a modulator of cellular metabolism.
65. The method of claim 64, wherein the modulator of cellular metabolism comprises nutrient starvation, a chemical inhibitor, or a small molecule.
66. The method of claim 65, wherein the chemical inhibitor or the small molecule is an mTOR inhibitor.
67. The method of claim 66, wherein the mTOR inhibitor comprises rapamycin or a derivative thereof.
68. A composition comprising one or more mitochondria-replaced cells obtained by:
(a) contacting a recipient cell with an agent that reduces the copy number of endogenous mtDNA;
(b) incubating the recipient cell with the agent for a sufficient period of time to partially reduce the endogenous mtDNA copy number in the recipient cell; and
(c) co-incubating (1) the recipient cell from step (b) in which the endogenous mtDNA has been partially depleted and (2) an exogenous mitochondrion from a healthy donor for a period of time sufficient to non-invasively transfer the exogenous mitochondrion to the recipient cell, thereby producing a mitochondrion-replaced cell,
wherein the mitochondrially replaced cell comprises greater than 5% exogenous mtDNA.
69. A composition of one or more mitochondria-replaced cells obtained by:
(a) contacting a recipient cell with an agent that reduces the copy number of endogenous mtDNA;
(b) incubating the recipient cell with the agent for a sufficient period of time to partially reduce the endogenous mtDNA copy number in the recipient cell; and
(c) co-incubating (1) the recipient cell from step (b) in which endogenous mtDNA has been partially reduced and (2) exogenous mtDNA from a healthy donor for a period of time sufficient to non-invasively transfer exogenous mtDNA to the recipient cell, thereby producing a mitochondria-replaced cell,
wherein the mitochondrially replaced cell comprises greater than 5% exogenous mtDNA.
70. The composition of claim 68 or 69, wherein the one or more mitochondria-replaced cells comprise a total mtDNA copy number of no more than about 1.1-fold, about 1.2-fold, about 1.3-fold, about 1.4-fold, about 1.5-fold, or more, relative to the total mtDNA copy number of the recipient cell prior to contact with the agent that reduces the endogenous mtDNA copy number.
71. A composition for use in a method of producing one or more mitochondrial-replaced cells, the composition comprising an agent that reduces endogenous mtDNA copy number and a second active agent.
72. The composition of claim 71, further comprising one or more recipient cells or a combination thereof.
73. The composition of claim 71 or 72, further comprising exogenous mtDNA, exogenous mtDNA and/or exogenous mitochondria.
74. The composition of any one of claims 68-73, wherein the agent that reduces endogenous mtDNA copy number is a small molecule or a fusion protein.
75. The composition of claim 74, wherein the small molecule is a Nucleoside Reverse Transcriptase Inhibitor (NRTI).
76. The composition of claim 74, wherein the fusion protein comprises an mtDNA cleaving endonuclease and a Mitochondrial Targeting Sequence (MTS).
77. The composition of claim 76, wherein said endonuclease cleaves wild-type mtDNA.
78. The composition of claim 77, wherein the endonuclease is selected from XbaI, EcoRI, BamHI, HindIII, PstI, Cas9, Zinc Finger Nuclease (ZFN), and transcription activator-like effector nuclease (TALEN).
79. The composition according to any one of claims 76-78, wherein the MTS is targeted to a mitochondrial matrix protein.
80. The composition of claim 79, wherein the mitochondrial matrix protein is selected from cytochrome c oxidase subunit IV, cytochrome c oxidase subunit VIII, and cytochrome c oxidase subunit X.
81. The composition of any one of claims 74-80, wherein the fusion protein is transiently expressed.
82. The composition of any one of claims 68-81, wherein said reduction in endogenous mtDNA copy number is a partial reduction.
83. The composition of claim 82, wherein said partial reduction is a reduction in endogenous mtDNA of about 5% to about 99%.
84. The composition of claim 82, wherein said partial reduction is a reduction in the copy number of said endogenous mtDNA of about 50% to about 95%.
85. The composition of claim 82, wherein said partial reduction is a reduction in the copy number of said endogenous mtDNA of about 60% to about 90%.
86. A composition comprising one or more mitochondria-replaced cells obtained by:
(a) contacting the recipient cell with an agent that reduces mitochondrial function;
(b) incubating the recipient cell with the agent for a period of time sufficient to partially reduce endogenous mitochondrial function in the recipient cell; and
(c) co-incubating (1) the recipient cell from step (b) in which endogenous mitochondrial function has been partially reduced and (2) exogenous mitochondria from a healthy donor for a period of time sufficient to non-invasively transfer the exogenous mitochondria to the recipient cell, thereby producing a mitochondria-replaced cell,
Wherein the mitochondrially replaced cell comprises greater than 5% exogenous mtDNA.
87. A composition of one or more mitochondria-replaced cells obtained by:
(a) contacting the recipient cell with an agent that reduces mitochondrial function;
(b) incubating the recipient cell with the agent for a period of time sufficient to partially reduce endogenous mitochondrial function in the recipient cell; and
(c) co-incubating (1) the recipient cell from step (b) in which endogenous mitochondrial function has been partially diminished and (2) exogenous mtDNA from a healthy donor for a period of time sufficient to non-invasively transfer the exogenous mtDNA to the recipient cell, thereby producing a mitochondrial replaced cell,
wherein the mitochondrially replaced cell comprises greater than 5% exogenous mtDNA.
88. The composition of claim 86 or 87, wherein the one or more mitochondria-replaced cells comprise a total mtDNA copy number of no more than about 1.1-fold, about 1.2-fold, about 1.3-fold, about 1.4-fold, about 1.5-fold, or more, relative to the total mtDNA copy number of the recipient cell prior to contact with the agent that reduces the endogenous mtDNA copy number.
89. A composition for use in a method of producing one or more mitochondria-replaced cells, the composition comprising an agent that reduces mitochondrial function and a second active agent.
90. The composition of claim 89, further comprising an exogenous mitochondrion, one or more recipient cells, or a combination thereof.
91. The composition of claim 89 or 90, further comprising exogenous mtDNA.
92. The composition of claim 68 or 69, wherein the one or more mitochondrially-replaced cells comprise wild-type exogenous mtDNA.
93. The composition of claim 68 or 69, further comprising a second active agent.
94. The composition of claim 71 or 93, wherein the second active agent is selected from a macromolecule, a small molecule, or a cell therapy, and the second active agent is optionally selected from rapamycin, NR (nicotinamide ribose), bezafibrate, idebenone, mercaptoethylamine bitartrate (RP103), elamipramide (MTP131), omavirulone (omavelolone) (RTA408), KH176, vantiquinone (Vatiquinone) (Epi743), lipoic acid, a0001 (alpha-tocopherolquinone), mitochondrial CoQ10(MitoQ), SkQ1(Visomitin), resveratrol, curcumin, ketonic therapy, hypoxia, and an endocytic activator.
95. The composition of claim 94, wherein said endocytic activator is an activator of an endostatin-dependent endocytic pathway.
96. The composition of claim 95, wherein said endocytic activator is an activator of an endostatin-dependent endocytic pathway.
97. The composition of claim 96, wherein the endostatin-dependent endocytosis pathway is selected from the group consisting of CLIC/GEEC endocytosis pathway, Arf 6-dependent endocytosis, lipocalin-dependent endocytosis, macroendocytosis, circular membrane ruffles (circular membrane ruffles), phagocytosis, and trans-endocytosis.
98. The composition of claim 96, wherein the endostatin-dependent endocytic pathway is megapinocytosis.
99. The composition of claim 95, wherein said endocytic activator comprises a nutritional stress and/or an mTOR inhibitor.
100. The composition of claim 99, wherein the mTOR inhibitor comprises rapamycin or a derivative thereof.
101. The composition of any one of claims 68-100, wherein the total mtDNA copy number of the one or more mitochondrially-replaced cells comprises greater than 5% exogenous mtDNA.
102. The composition of any one of claims 68-101, wherein the total mtDNA copy number of the one or more mitochondrially-replaced cells comprises greater than 30% exogenous mtDNA.
103. The composition of any one of claims 68-102, wherein the total mtDNA copy number of the one or more mitochondrially-replaced cells comprises greater than 50% exogenous mtDNA.
104. The composition of any one of claims 68-103, wherein the total mtDNA copy number of the one or more mitochondrially-replaced cells comprises greater than 75% exogenous mtDNA.
105. The composition of claim 68, wherein the exogenous mitochondria is an isolated mitochondria.
106. The composition of claim 105, wherein the isolated mitochondria are intact.
107. The composition of any one of claims 68-106, wherein said exogenous mitochondrial and/or exogenous mtDNA is allogeneic.
108. The composition of claim 68, wherein the exogenous mitochondria further comprise exogenous mtDNA.
109. The composition of any one of claims 68-108, wherein the one or more cells are animal cells or plant cells.
110. The composition of claim 109, wherein the animal cell is a mammalian cell.
111. The composition of claim 110, wherein the cell is a somatic cell.
112. The composition of claim 111, wherein the somatic cell is an epithelial cell.
113. The composition of claim 112, wherein the epithelial cells are Thymic Epithelial Cells (TEC).
114. The composition of claim 111, wherein the somatic cell is an immune cell.
115. The composition of claim 114, wherein the immune cell is a T cell.
116. The composition of claim 115, wherein the T cell is a CD4+ T cell.
117. The composition of claim 115, wherein the T cell is a CD8+ T cell.
118. The composition of claim 115, wherein the T cell is a Chimeric Antigen Receptor (CAR) T cell.
119. The composition of claim 114, wherein the immune cell is a phagocytic cell.
120. The composition of any one of claims 68-108, wherein the one or more mitochondria-replaced cells are bone marrow cells.
121. The composition of claim 120, wherein the bone marrow cells are Hematopoietic Stem Cells (HSCs) or Mesenchymal Stem Cells (MSCs).
122. The composition of any one of claims 68-121, wherein the one or more mitochondria-replaced cells are more viable than isogenic cells having homogeneous endogenous mtDNA.
123. The composition of any one of claims 68-122, wherein the one or more mitochondria replaced cells are effective in killing cancer cells, treating an age-related disease, treating a mitochondrial disease or disorder, treating a neurodegenerative disease, treating diabetes, or a genetic disease.
124. The composition of any one of claims 68-123, further comprising a small molecule, peptide, or protein.
125. A composition for delaying senescence and/or extending lifespan in a cell, comprising:
(a) senescent or near-senescent cells with endogenous mitochondria;
(b) isolated exogenous mitochondria from non-senescent cells; and
(c) an agent that reduces the copy number of endogenous mtDNA.
126. The composition of claim 125, wherein the agent is a fusion protein.
127. The composition of claim 126, wherein the fusion protein comprises an mtDNA cleaving endonuclease and a Mitochondrial Targeting Sequence (MTS).
128. The composition of claim 127, wherein said endonuclease cleaves wild-type mtDNA.
129. The composition of claim 127 or 128, wherein the endonuclease is selected from XbaI, EcoRI, BamHI, HindIII, PstI, Cas9, Zinc Finger Nuclease (ZFN), and transcription activator-like effector nuclease (TALEN).
130. The composition of any one of claims 127-129, wherein the MTS targets a mitochondrial matrix protein.
131. The composition of claim 130, wherein the mitochondrial matrix protein is selected from cytochrome c oxidase subunit IV, cytochrome c oxidase subunit VIII, and cytochrome c oxidase subunit X.
132. The composition of any one of claims 126-131, wherein the fusion protein is transiently expressed in the senescent or near-senescent cell.
133. A composition for delaying senescence and/or extending lifespan in a cell, comprising:
(a) senescent or near-senescent cells with endogenous mitochondria;
(b) isolated exogenous mitochondria from non-senescent cells; and
(c) an agent that reduces mitochondrial function.
134. The composition of claim 133, wherein the agent that reduces mitochondrial function transiently reduces endogenous mitochondrial function.
135. The composition of claim 133, wherein the agent that reduces mitochondrial function permanently reduces endogenous mitochondrial function.
136. The composition of any one of claims 125-135, wherein the exogenous mitochondria from the non-senescent cell have increased function relative to the endogenous mitochondria.
137. The composition of any one of claims 125-132, further comprising a second active agent.
138. The composition of claim 137, wherein the second active agent is selected from a macromolecule, a small molecule, or a cell therapy, and the second active agent is optionally selected from rapamycin, NR (nicotinamide ribose), bezafibrate, idebenone, mercaptoethylamine bitartrate (RP103), elamipramide (MTP131), omaviralone (omavelolone) (RTA408), KH176, vatilquinone (Vatiquinone) (Epi743), lipoic acid, a0001 (alpha-tocopherolquinone), mitochondrial CoQ10(MitoQ), SkQ1(Visomitin), resveratrol, curcumin, a ketonic therapy, hypoxia, and an endocytic activator.
139. The composition of claim 138, wherein said endocytic activator is an activator of an endostatin-dependent endocytic pathway.
140. The composition of claim 139, wherein the endostatin-dependent endocytosis pathway is selected from the group consisting of CLIC/GEEC endocytosis pathway, Arf 6-dependent endocytosis, lipocalin-dependent endocytosis, macroendocytosis, circular membrane ruffles (circular nuclear ruffles), phagocytosis, and trans-endocytosis.
141. The composition of claim 139, wherein the endostatin-dependent endocytic pathway is megapinocytosis.
142. The composition of claim 138, wherein said endocytic activator comprises a nutritional stress and/or an mTOR inhibitor.
143. The composition of claim 142, wherein the mTOR inhibitor comprises rapamycin or a derivative thereof.
144. A pharmaceutical composition comprising an isolated population of cells having mitochondrial replacement of an exogenous mitochondrion from a healthy donor, wherein the cells are obtained by the method of claim 1, 26, or 62.
145. A pharmaceutical composition comprising an isolated population of cells having mitochondrial replacement of exogenous mtDNA from a healthy donor, wherein the cells are obtained by the method of claim 1, 10, 26, 27, or 62.
146. The pharmaceutical composition of claim 144, further comprising an exogenous mitochondrion.
147. The pharmaceutical composition of any one of claims 144-146, further comprising a pharmaceutically acceptable carrier.
148. The pharmaceutical composition of any one of claims 144-147, wherein the cell is a T cell.
149. The pharmaceutical composition of any one of claims 144-147, wherein the cells are hematopoietic stem cells.
1. Field of the invention
The present invention provides compositions of cells with reduced mitochondrial DNA and/or mitochondrial DNA replacement, methods for producing them, and methods for treating various diseases associated with genetic or age-related mitochondrial dysfunction.
2. Background of the invention
Mitochondria play an important and critical role in cellular homeostasis and are involved in a wide range of disease processes. They are involved in intracellular signal transduction, apoptosis and perform multiple biochemical tasks such as pyruvate oxidation, the tricarboxylic acid cycle and amino acid, fatty acid, nucleotide and steroid metabolism. One key task is their role in cellular energy metabolism. This includes β -fatty acid oxidation and ATP production by the electron transport chain and oxidation-phosphorylation system. The mitochondrial respiratory chain consists of a complex of 5 multi-subunit proteins embedded in the inner membrane, which includes: complex I (NADH-ubiquinone oxidoreductase), complex II (succinate-ubiquinone oxidoreductase), complex III (ubiquinol-ferrocytochrome c oxidoreductase), complex IV (cytochrome c oxidoreductase) and complex V (FIFO atpase).
The mammalian mitochondrial genome is a small, circular, double-stranded molecule containing 37 genes, which includes 13 protein-encoding genes, 22 transfer rna (trna) genes, and 2 ribosomal rna (rrna) genes. Of these, mitochondrial DNA translation requires 24 (22 trnas and 2 rrnas), and 13 subunits encoding the respiratory chain complex. In addition, nuclear dna (ndna) encodes the majority of about 900 gene products in mitochondria.
Mitochondrial diseases or disorders are a clinically heterogeneous group of disorders characterized by dysfunctional mitochondria. Disease onset can occur at any age and can exhibit a wide range of clinical symptoms. Mitochondrial diseases or disorders may involve any organ or tissue, which characteristically involves multiple systems, often affecting organs that are highly dependent on aerobic metabolism and often develop crudely with high morbidity and mortality. Mitochondrial diseases or disorders are the most common group of inherited metabolic disorders, and the most common form of inherited neurological disorders.
Mitochondrial diseases or disorders can be caused by genetic mutations in nuclear dna (ndna) and/or mitochondrial dna (mtdna) encoding structural mitochondrial proteins or proteins involved in mitochondrial function. Although some mitochondrial disorders affect only a single organ (e.g., in Leber's hereditary optic neuropathy LHON affecting the eye), a variety of disorders involve multiple organ systems and exist with distinct neurological and myopic features. Although more frequently involving tissues with high energy requirements, such as the brain, muscles and eyes, the phenotype of patients can be very different and heterogeneous. This difference is due in part to several factors such as dual genetic control (nDNA and mtDNA) levels of heterogeneity (percentage of mutant DNA in single cells and tissues), tissue energy requirements, maternal inheritance and mitotic segregation.
Multiple patients with mitochondrial diseases or disorders have a mixture of mutant and wild-type mtDNA (referred to as heterogeneity); the ratio of mutant and wild type mtDNA is a key factor in determining whether a cell expresses a biochemical defect. Most pathogenic mtDNA mutations are heterogeneous, with a mix of mutant and wild-type mtDNA within each cell. A high level of heterogeneity indicates cells with high levels of mutant mtDNA and low levels of wild-type mtDNA, while a low level of heterogeneity indicates cells with low levels of mutant mtDNA and high levels of wild-type mtDNA. Studies in single cells from patients with mitochondrial diseases or disorders have shown that the levels of mutant and wild-type mtDNA are very important in determining the phenotype of the cell. For example, if they contain high levels of mutant mtDNA and low levels of wild-type mtDNA (i.e., high levels of heterogeneity), the cells become respiratory deficient. The threshold at which this defect occurs depends on the exact mutation and cell type. Typically, high mutant mtDNA percentage levels (> 50%) are required to produce cellular defects, but some mtDNA mutations only produce defects if present at very high levels (typically mt tRNA mutations), while others (e.g., single, large-scale mtDNA deletions) produce defects when-60% of the deleted mtDNA is present. For example, in individuals with the m.8993T > G pathogenic variant, higher percent levels of mutant mtDNA were observed in those with Leigh syndrome than in those with neurogenic weakness with ataxia and retinitis pigmentosa (NARP). In addition, the clinical phenotypes in MELAS and MERRF are associated with heterogeneity (see, e.g., Chinnery, P.F. et al, Brain 120(Pt 10),1713-1721 (1997)).
The development of next generation sequencing technologies has shown a variety of mutations that cause mitochondrial diseases or disorders. In addition, studies of other organisms, such as caenorhabditis elegans (c.elegans), have shown that some proteins are involved in heterogeneity. For example, recent studies using C.elegans have shown that following perturbation of the original mtDNA, the mitochondrial unfolded protein reaction (UPRmt) acts to maintain heterogeneity and spread the mutant mtDNA (see, e.g., Lin, Y.F. et al, Nature 533,416-419, doi:10.1038/Nature17989 (2016)). However, the mechanisms associated with the maintenance and spread of heterogeneity in mammalian cells are still unknown.
Control and treatment of patients with mitochondrial diseases or disorders remains difficult. For the vast majority of patients, the condition progresses crudely, leading to high morbidity and, among those most affected, death. Typical methods for removal of endogenous mtDNA include long-term treatment of cells with low concentrations of ethidium bromide (EtBr, a known carcinogen and teratogen), thereby limiting their use for therapeutic purposes. In addition to the potential for undesirable side effects, the EtBr protocol can take months, which further limits its clinical use. In addition, mitochondrial transfer protocols typically involve the complete elimination of endogenous mtDNA prior to external source mitochondrial transfer, referred to as rho (ρ)0 cells. This complete elimination of mtDNA severely hampers the ability of cells to take up exogenous mitochondria.
Other mitochondrial transfer protocols have been attempted to add mitochondria without eliminating endogenous mtDNA, but this approach has been found to be ineffective or detrimental to cells. For example, mitochondrial transfer using simple co-incubation has been reported to be ineffective and not as effective in different cell types. Other transfer techniques have included infusion using invasive instruments that are harmful to recipient Cells, or the use of other invasive instruments such as nanoplatelets, none of which are as effective as co-incubation (caicido et al, Stem Cells International, (2017), volume 2017, paper ID 7610414, page 23).
Thus, not only are current methods of mitochondrial transfer impractical for clinical settings, they are also inefficient, detrimental to recipient cells and/or time intensive. Thus, there remains a significant unmet need for the development of improved methods for mitochondrial metastasis and improved models for the study of mitochondrial diseases or disorders that can optionally be used in the treatment of subjects having or suspected of having mitochondrial diseases or disorders and diseases or disorders associated with damaged or dysfunctional mitochondria.
3. Summary of the invention
In one aspect, provided herein is a method of producing a mitochondrial-replaced cell comprising: (a) contacting a recipient cell with an agent that reduces the copy number of endogenous mtDNA; (b) incubating the recipient cell with the agent for a sufficient period of time to partially reduce the endogenous mtDNA copy number in the recipient cell; and (c) co-incubating (1) the recipient cell from step (b) in which endogenous mtDNA has been partially reduced and (2) exogenous mitochondria from a healthy donor for a period of time sufficient to non-invasively transfer the exogenous mitochondria to the recipient cell, thereby producing a mitochondria-replaced cell.
In another aspect, provided herein is a method of treating a subject in need of mitochondrial replacement comprising (a) generating ex vivo or in vitro a mitochondrial replaced cell comprising the steps of: (i) contacting a recipient cell with an agent that reduces mtDNA copy number; (ii) incubating the recipient cell with the agent for a period of time sufficient to partially reduce mtDNA copy number in the recipient cell; and (iii) co-incubating (1) the recipient cell from step (ii) in which endogenous mtDNA has been partially reduced and (2) exogenous mitochondria from a healthy donor for a sufficient period of time to non-invasively transfer exogenous mitochondria to the recipient cell, thereby producing a mitochondria-replaced cell; and (b) administering a therapeutically effective amount of the receptor cell for mitochondrial replacement from step (a) to a subject in need of mitochondrial replacement.
In another aspect, provided herein is a method of treating a subject having or suspected of having an age-related disease, the method comprising: (a) producing ex vivo or in vitro a mitochondrial replaced cell comprising the steps of: (i) contacting a recipient cell with an agent that reduces mtDNA copy number; (ii) incubating the recipient cell with the agent for a period of time sufficient to partially reduce mtDNA copy number in the recipient cell; and (iii) co-incubating (1) the recipient cell from step (ii) in which endogenous mtDNA has been partially reduced and (2) exogenous mitochondria from a healthy donor for a sufficient period of time to non-invasively transfer exogenous mitochondria to the recipient cell, thereby producing a mitochondria-replaced cell; and (b) administering a therapeutically effective amount of the mitochondrially-replaced recipient cell from step (a) to a subject having or suspected of having an age-related disorder.
In another aspect, provided herein is a method of treating a subject having or suspected of having a mitochondrial disease or disorder, the method comprising: (a) ex vivo or in vitro production of a mitochondrially replaced recipient cell comprising the steps of: (i) contacting a recipient cell with an agent that reduces mtDNA copy number; (ii) incubating the recipient cell with the agent for a period of time sufficient to partially reduce mtDNA copy number in the recipient cell; and (iii) co-incubating (1) the recipient cell from step (ii) in which endogenous mtDNA has been partially reduced and (2) exogenous mitochondria from a healthy donor for a sufficient period of time to non-invasively transfer exogenous mitochondria to the recipient cell, thereby producing a mitochondria-replaced cell; and (b) administering a therapeutically effective amount of the mitochondrial replaced recipient cell from step (a) to a subject having or suspected of having a mitochondrial disease or disorder.
In some embodiments of the methods provided herein, the exogenous mitochondria are functional mitochondria. In certain embodiments, the exogenous mitochondrion comprises wild-type mtDNA. In a specific embodiment, the exogenous mitochondria is an isolated mitochondria. In other embodiments, the isolated mitochondria are intact mitochondria. In some embodiments, the exogenous mitochondria are allogeneic.
Also provided herein are methods of producing a mitochondrial-replaced cell comprising: (a) contacting a recipient cell with an agent that reduces the copy number of endogenous mtDNA; (b) incubating the recipient cell with the agent for a sufficient period of time to partially reduce the endogenous mtDNA copy number in the recipient cell; and (c) co-incubating (1) the recipient cell from step (b) in which the endogenous mtDNA has been partially depleted and (2) the exogenous mtDNA from the healthy donor for a period of time sufficient to non-invasively transfer the exogenous mtDNA to the recipient cell, thereby producing a mitochondria-replaced cell.
The present disclosure also provides a method of treating a subject in need of mitochondrial replacement comprising: (a) producing ex vivo or in vitro a mitochondrial replaced cell comprising the steps of: (i) contacting a recipient cell with an agent that reduces mtDNA copy number (ii) incubating the recipient cell with the agent for a sufficient period of time to partially reduce mtDNA copy number in the recipient cell; and (iii) co-incubating (1) the recipient cell from step (ii) in which endogenous mtDNA has been partially reduced and (2) exogenous mtDNA from a healthy donor for a period of time sufficient to non-invasively transfer exogenous mtDNA to the recipient cell, thereby producing a mitochondria-replaced cell; and (b) administering a therapeutically effective amount of the receptor cell for mitochondrial replacement from step (a) to a subject in need of mitochondrial replacement.
In another aspect, provided herein is a method of treating a subject having or suspected of having an age-related disease, the method comprising: (a) producing ex vivo or in vitro a mitochondrial replaced cell comprising the steps of: (i) contacting a recipient cell with an agent that reduces mtDNA copy number; (ii) incubating the recipient cell with the agent for a period of time sufficient to partially reduce mtDNA copy number in the recipient cell; and (iii) co-incubating (1) the recipient cell from step (ii) in which endogenous mtDNA has been partially reduced and (2) exogenous mtDNA from a healthy donor for a period of time sufficient to non-invasively transfer exogenous mtDNA to the recipient cell, thereby producing a mitochondria-replaced cell; and (b) administering a therapeutically effective amount of the mitochondrially-replaced recipient cell from step (a) to a subject having or suspected of having an age-related disorder.
In another aspect, provided herein is a method of treating a subject having or suspected of having a mitochondrial disease or disorder, the method comprising: (a) ex vivo or in vitro production of a mitochondrially replaced recipient cell comprising the steps of: (i) contacting a recipient cell with an agent that reduces mtDNA copy number; (ii) incubating the recipient cell with the agent for a period of time sufficient to partially reduce mtDNA copy number in the recipient cell; and (iii) co-incubating (1) the recipient cell from step (ii) in which endogenous mtDNA has been partially reduced and (2) exogenous mtDNA from a healthy donor for a period of time sufficient to non-invasively transfer exogenous mtDNA to the recipient cell, thereby producing a mitochondria-replaced cell; and (b) administering a therapeutically effective amount of the mitochondrial replaced recipient cell from step (a) to a subject having or suspected of having a mitochondrial disease or disorder.
In certain embodiments of the methods provided herein, the agent that reduces the copy number of endogenous mtDNA is selected from the group consisting of a polynucleotide encoding a fusion protein comprising a mitochondrial-targeting sequence (MTS) and an endonuclease, a polynucleotide encoding an endonuclease, and a small molecule. In some embodiments, the small molecule is a Nucleoside Reverse Transcriptase Inhibitor (NRTI). In other embodiments, the polynucleotide consists of a messenger ribonucleic acid (mRNA) or a deoxyribonucleic acid (DNA). In other embodiments, the recipient cell transiently expresses the fusion protein. In other embodiments, the endonuclease is selected from the group consisting of XbaI, EcoRI, BamHI, HindIII, PstI, Cas9, Zinc Finger Nuclease (ZFN), and transcription activator-like effector nuclease (TALEN). In some embodiments, the MTS targets mitochondrial matrix proteins. In a specific embodiment, the mitochondrial matrix protein is selected from cytochrome c oxidase subunit IV, cytochrome c oxidase subunit VIII, and cytochrome c oxidase subunit X.
In some embodiments of the methods provided herein, the agent that reduces endogenous mtDNA copy number by about 5% to about 99%. In certain embodiments, the agent that reduces endogenous mtDNA copy number by about 30% to about 70%. In other embodiments, the agent that reduces endogenous mtDNA copy number by about 50% to about 95%. In other embodiments, the agent that reduces endogenous mtDNA copy number by about 60% to about 90%. In some embodiments, the agent that reduces the copy number of endogenous mtDNA reduces mitochondrial material.
Also provided herein are methods of producing a mitochondrial-replaced cell comprising: (a) contacting the recipient cell with an agent that reduces mitochondrial function; (b) incubating the recipient cell with the agent for a period of time sufficient to partially reduce endogenous mitochondrial function in the recipient cell; and (c) co-incubating (1) the recipient cell from step (b) in which endogenous mitochondrial function has been partially reduced and (2) exogenous mitochondria from a healthy donor for a period of time sufficient to non-invasively transfer the exogenous mitochondria to the recipient cell, thereby producing a mitochondria-replaced cell.
The present disclosure also provides a method of producing a mitochondrial-replaced cell comprising: (a) contacting the recipient cell with an agent that reduces mitochondrial function; (b) incubating the recipient cell with the agent for a period of time sufficient to partially reduce endogenous mitochondrial function in the recipient cell; and (c) co-incubating (1) the recipient cell from step (b) in which endogenous mitochondrial function has been partially reduced and (2) exogenous mtDNA from a healthy donor for a period of time sufficient to non-invasively transfer the exogenous mtDNA to the recipient cell, thereby producing a mitochondrial-replaced cell.
In some embodiments of the methods provided herein, the agent that decreases mitochondrial function transiently decreases endogenous mitochondrial function. In other embodiments, the agent that reduces mitochondrial function permanently reduces endogenous mitochondrial function.
In certain embodiments of the methods provided herein, a subject in need of mitochondrial replacement has dysfunctional mitochondria; a disease selected from the group consisting of age-related diseases, mitochondrial diseases or disorders, neurodegenerative diseases, retinal diseases, diabetes, hearing disorders, genetic diseases; or a combination thereof. In some embodiments, the neurodegenerative disease is selected from Amyotrophic Lateral Sclerosis (ALS), huntington's disease, alzheimer's disease, parkinson's disease, friedrich's ataxia, peroneal muscular atrophy, and cerebral leukosis. In a specific embodiment, the retinal disease is selected from the group consisting of age-related macular degeneration, macular edema, and glaucoma.
In some embodiments of the methods provided herein, the age-related disease is selected from the group consisting of an autoimmune disease, a metabolic disease, a genetic disease, cancer, a neurodegenerative disease, and immunosenescence. In certain embodiments of the methods provided herein, the metabolic disease is diabetes. In other embodiments, the neurodegenerative disease is alzheimer's disease or parkinson's disease. In other embodiments, the genetic disease is selected from the group consisting of early aging syndrome, vorner syndrome, and huntington's disease.
In certain embodiments of the methods provided herein, the mitochondrial disease or disorder is caused by an abnormality in mitochondrial DNA, an abnormality in nuclear DNA, or both. In particular embodiments, the mitochondrial disease or disorder caused by mitochondrial DNA abnormalities is selected from Chronic Progressive External Ophthalmoplegia (CPEO), pearson Syndrome, cohns-Sayre Syndrome (KSS), diabetes with deafness (DAD), mitochondrial diabetes, Leber Hereditary Optic Neuropathy (LHON), LHON-plus (LHON-plus), neuropathy, ataxia and retinitis pigmentosa Syndrome (NARP), Maternally Inherited Leigh Syndrome (MILS), mitochondrial encephalomyopathy, lactic acidosis and stroke-like attacks (MELAS), myoclonic epilepsy with morbid red fiber disease (MERRF), familial bilateral striatal necrosis/striatal nigra degeneration (FBSN), Luft disease, aminoglycoside-induced deafness (AID), and various deletions of mitochondrial DNA Syndrome. In other embodiments, the mitochondrial disease or disorder caused by a nuclear DNA abnormality is selected from the group consisting of mitochondrial DNA deletion syndrome-4A, mitochondrial recessive ataxia syndrome (MIRAS), mitochondrial neurogastrointestinal encephalomyopathy (MNGIE), mitochondrial DNA deletion syndrome (MTDPS), DNA polymerase gamma (POLG) -associated disorders, sensory ataxia neuropathy with dysarthria and ophthalmoplegia (SANDO), cerebral leukosis with involvement of the brain stem and spinal cord and elevated lactate (LBSL), coenzyme Q10 deficiency, Leigh syndrome, mitochondrial complex abnormalities, fumarate deficiency, alpha-ketoglutardialdehyde dehydrogenase complex (KGDHC) deficiency, succinyl-coa ligase deficiency, pyruvate dehydrogenase complex deficiency (PDHC), Pyruvate Carboxylase Deficiency (PCD), carnitine palmitoyltransferase i cpt i) deficiency, and, Carnitine palmitoyltransferase ii (cpt ii) deficiency, carnitine-acyl-carnitine (CACT) deficiency, autosomal dominant-/autosomal recessive-progressive extra-ocular paralysis (ad-/ar-PEO), infantile spinocerebellar atrophy (IOSCA), Mitochondrial Myopathy (MM), Spinal Muscular Atrophy (SMA), growth arrest, aminouria, cholestasis, iron overload, early death (GRACILE), and Charcot-Marei-Tooth 2A (CMT 2A).
In some embodiments of the methods provided herein, the endogenous mtDNA encodes a dysfunctional mitochondrion. In a specific embodiment, the endogenous mtDNA comprises a mutant mtDNA. In other embodiments, the endogenous mtDNA in the recipient cell comprises wild-type mtDNA. In other embodiments, the endogenous mtDNA comprises mtDNA associated with a mitochondrial disease or disorder. In some embodiments, the endogenous mtDNA is heterogeneous. In a specific embodiment, the recipient cell has dysfunctional endogenous mitochondria.
In certain embodiments of the methods provided herein, the mitochondria-replaced cell has a total mtDNA copy number of no greater than about 1.1-fold, about 1.2-fold, about 1.3-fold, about 1.4-fold, about 1.5-fold, or more, relative to the total mtDNA copy number of the recipient cell prior to contact with the agent that reduces the endogenous mtDNA copy number.
In some embodiments, the recipient cell is an animal cell or a plant cell. In certain embodiments, the animal cell is a mammalian cell. In a specific embodiment, the recipient cell is a somatic cell. In other embodiments, the recipient cell is a bone marrow cell. In some embodiments, the bone marrow cells are Hematopoietic Stem Cells (HSCs) or Mesenchymal Stem Cells (MSCs). In other embodiments, the recipient cell is a cancer cell. In other embodiments, the recipient cell is a primary cell. In other embodiments, the recipient cell is an immune cell. In specific embodiments, the immune cell is selected from the group consisting of a T cell, a phagocyte, a microglia, and a macrophage. In other embodiments, the T cell is a CD4+ T cell. In other embodiments, the T cell is a CD8+ T cell. In certain embodiments, the T cell is a Chimeric Antigen Receptor (CAR) T cell.
In another embodiment of the methods provided herein, the exogenous mitochondrial and/or exogenous mtDNA is stable. In some embodiments, the exogenous mtDNA alters heterogeneity in the recipient cell.
In some aspects of the methods provided herein, the methods further comprise delivering a small molecule, peptide, or protein.
The present disclosure also provides methods provided herein, further comprising contacting the recipient cell with a second active agent prior to co-incubating the recipient cell with exogenous mitochondria and/or exogenous mtDNA. In certain embodiments, the second active agent is selected from a macromolecule, a small molecule, or a cell therapy, and the second active agent is optionally selected from rapamycin, NR (nicotinamide ribose), bezafibrate, idebenone, mercaptoethylamine bitartrate (RP103), elamipramide (MTP131), omaviralone (omavelolone) (RTA408), KH176, vantiquone (Vatiquinone) (Epi743), lipoic acid, a0001 (alpha-tocopherols), mitochondrial CoQ10(MitoQ), SkQ1(Visomitin), resveratrol, curcumin, ketonetherapy, hypoxia, and an endocytosis activator. In some embodiments, the endocytic activator is a modulator of cellular metabolism. In particular embodiments, the modulator of cellular metabolism comprises nutrient starvation, a chemical inhibitor, or a small molecule. In other embodiments, the chemical inhibitor or the small molecule is an mTOR inhibitor. In other embodiments, the mTOR inhibitor comprises rapamycin or a derivative thereof.
The present disclosure also provides compositions comprising one or more mitochondria-replaced cells obtained by: (a) contacting a recipient cell with an agent that reduces the copy number of endogenous mtDNA; (b) incubating the recipient cell with the agent for a sufficient period of time to partially reduce the endogenous mtDNA copy number in the recipient cell; and (c) co-incubating (1) the recipient cell from step (b) in which the endogenous mtDNA has been partially depleted and (2) an exogenous mitochondrion from a healthy donor for a period of time sufficient to non-invasively transfer the exogenous mitochondrion to the recipient cell, thereby producing a mitochondrion-replaced cell, wherein the mitochondrion-replaced cell comprises greater than 5% exogenous mtDNA.
The present disclosure also provides compositions of one or more mitochondria-replaced cells obtained by: (a) contacting a recipient cell with an agent that reduces the copy number of endogenous mtDNA; (b) incubating the recipient cell with the agent for a sufficient period of time to partially reduce the endogenous mtDNA copy number in the recipient cell; and (c) co-incubating (1) the recipient cell from step (b) in which endogenous mtDNA has been partially reduced and (2) exogenous mtDNA from a healthy donor for a period of time sufficient to non-invasively transfer exogenous mtDNA to the recipient cell, thereby producing a mitochondria-replaced cell, wherein the mitochondria-replaced cell comprises greater than 5% exogenous mtDNA. In some embodiments of the compositions provided herein, the one or more mitochondria-replaced cells comprise a total mtDNA copy number of no greater than about 1.1-fold, about 1.2-fold, about 1.3-fold, about 1.4-fold, about 1.5-fold, or more, relative to the total mtDNA copy number of the recipient cell prior to contact with the agent that reduces the endogenous mtDNA copy number.
In another aspect, provided herein are compositions for use in methods of producing one or more mitochondrial replaced cells comprising an agent that reduces endogenous mtDNA copy number and a second active agent. In some embodiments, the composition further comprises one or more recipient cells or a combination thereof. In certain embodiments, the composition further comprises exogenous mtDNA, and/or exogenous mitochondria.
In certain embodiments of the compositions provided herein, the agent that reduces the copy number of endogenous mtDNA is a small molecule or a fusion protein. In some embodiments, the small molecule is a Nucleoside Reverse Transcriptase Inhibitor (NRTI). In other embodiments, the fusion protein comprises an mtDNA cleaving endonuclease and a Mitochondrial Targeting Sequence (MTS). In some embodiments, the endonuclease cleaves wild-type mtDNA. In specific embodiments, the endonuclease is selected from the group consisting of XbaI, EcoRI, BamHI, HindIII, PstI, Cas9, Zinc Finger Nuclease (ZFN), and transcription activator-like effector nuclease (TALEN). In some embodiments, the MTS targets a mitochondrial matrix protein. In other embodiments, the mitochondrial matrix protein is selected from cytochrome c oxidase subunit IV, cytochrome c oxidase subunit VIII, and cytochrome c oxidase subunit X. In a specific embodiment, the fusion protein is transiently expressed.
In some embodiments of the compositions provided herein, the reduction in endogenous mtDNA copy number is a partial reduction. In certain embodiments, the partial reduction is a reduction in endogenous mtDNA of about 5% to about 99%. In a specific embodiment, the partial reduction is a reduction in the copy number of endogenous mtDNA of about 50% to about 95%. In other embodiments, the partial reduction is a reduction in the copy number of endogenous mtDNA of about 60% to about 90%.
The present disclosure also provides compositions comprising one or more mitochondria-replaced cells obtained by: (a) contacting the recipient cell with an agent that reduces mitochondrial function; (b) incubating the recipient cell with the agent for a period of time sufficient to partially reduce endogenous mitochondrial function in the recipient cell; and (c) co-incubating (1) the recipient cell from step (b) in which endogenous mitochondrial function has been partially reduced and (2) an exogenous mitochondrion from a healthy donor for a period of time sufficient to non-invasively transfer the exogenous mitochondrion to the recipient cell, thereby producing a mitochondrion-replaced cell, wherein the mitochondrion-replaced cell comprises greater than 5% exogenous mtDNA.
In another aspect, provided herein is a composition of one or more mitochondria-replaced cells obtained by: (a) contacting the recipient cell with an agent that reduces mitochondrial function; (b) incubating the recipient cell with the agent for a period of time sufficient to partially reduce endogenous mitochondrial function in the recipient cell; and (c) co-incubating (1) the recipient cell from step (b) in which endogenous mitochondrial function has been partially diminished and (2) exogenous mtDNA from a healthy donor for a period of time sufficient to non-invasively transfer the exogenous mtDNA to the recipient cell, thereby producing a mitochondria-replaced cell, wherein the mitochondria-replaced cell comprises greater than 5% exogenous mtDNA. In some embodiments, the one or more mitochondria-replaced cells comprise a total mtDNA copy number of no greater than about 1.1-fold, about 1.2-fold, about 1.3-fold, about 1.4-fold, about 1.5-fold, or more relative to the total mtDNA copy number of the recipient cell prior to contact with the agent that reduces the endogenous mtDNA copy number.
The present disclosure also provides compositions for use in methods of producing one or more mitochondrial-displaced cells, the compositions comprising an agent that reduces mitochondrial function and a second active agent. In some embodiments, the composition further comprises an exogenous mitochondrion, one or more recipient cells, or a combination thereof. In other embodiments, the composition further comprises exogenous mtDNA.
In some embodiments of the compositions provided herein, the one or more mitochondrial replaced cells comprise wild-type exogenous mtDNA.
Also provided herein are compositions further comprising a second active agent. In some embodiments, the second active agent is selected from a macromolecule, a small molecule, or a cell therapy, and the second active agent is optionally selected from rapamycin, NR (nicotinamide ribose), bezafibrate, idebenone, mercaptoethylamine bitartrate (RP103), elamipramide (MTP131), omaviralone (omavelolone) (RTA408), KH176, vantiquone (Vatiquinone) (Epi743), lipoic acid, a0001 (alpha-tocopherols), mitochondrial CoQ10(MitoQ), SkQ1(Visomitin), resveratrol, curcumin, a ketonic therapy, hypoxia, and an endocytic activator. In particular embodiments, the endocytic activator is an activator of an endostatin-dependent endocytic pathway. In some embodiments, the endocytic activator is an activator of an endostatin-dependent endocytic pathway. In other embodiments, the endostatin-dependent endocytosis pathway is selected from the group consisting of CLIC/GEEC endocytosis pathway, Arf 6-dependent endocytosis, lipocalin-dependent endocytosis, macropinocytosis, circular membrane ruffles (circular nuclear ruffles), phagocytosis, and trans-endocytosis. In other embodiments, the endostatin-dependent endocytic pathway is megapinocytosis. In a specific embodiment, the endocytic activator comprises a nutritional threat and/or an mTOR inhibitor. In some embodiments, the mTOR inhibitor comprises rapamycin or a derivative thereof.
In certain embodiments, the disclosure also provides compositions wherein the total mtDNA copy number of the one or more mitochondria-replaced cells comprises greater than 5% exogenous mtDNA. In some embodiments, the total mtDNA copy number of the one or more mitochondria-replaced cells comprises greater than 30% exogenous mtDNA. In a specific embodiment, the total mtDNA copy number of the one or more mitochondria-replaced cells comprises greater than 50% exogenous mtDNA. In other embodiments, the total mtDNA copy number of the one or more mitochondria-replaced cells comprises greater than 75% exogenous mtDNA.
In some embodiments of the compositions provided herein, the exogenous mitochondria are isolated mitochondria. In a specific embodiment, the isolated mitochondria are intact. In some embodiments, the exogenous mitochondrial and/or exogenous mtDNA is allogeneic. In a specific embodiment, the exogenous mitochondrion further comprises exogenous mtDNA.
In certain embodiments of the compositions provided herein, the one or more cells are animal cells or plant cells. In some embodiments, the animal cell is a mammalian cell. In a specific embodiment, the cell is a somatic cell. In other embodiments, the somatic cell is an epithelial cell. In other embodiments, the epithelial cells are Thymic Epithelial Cells (TECs). In other embodiments, the somatic cell is an immune cell. In certain embodiments, the immune cell is a T cell. In a specific embodiment, the T cell is a CD4+ T cell. In other embodiments, the T cell is a CD8+ T cell. In some embodiments, the T cell is a Chimeric Antigen Receptor (CAR) T cell. In other embodiments, the immune cell is a phagocytic cell. In certain embodiments, the one or more mitochondria-replaced cells are bone marrow cells. In specific embodiments, the bone marrow cells are Hematopoietic Stem Cells (HSCs) or Mesenchymal Stem Cells (MSCs).
In some embodiments of the compositions provided herein, the one or more mitochondrial replaced cells are more viable than isogenic cells with homogeneous endogenous mtDNA. In other embodiments, the one or more mitochondria replaced cells are effective in killing cancer cells, treating an age-related disease, treating a mitochondrial disease or disorder, treating a neurodegenerative disease, treating diabetes, or a genetic disease.
In certain embodiments of the compositions provided herein, the composition further comprises a small molecule, peptide, or protein.
Also provided herein are compositions for delaying senescence and/or increasing longevity in a cell, comprising: (a) senescent or near-senescent cells with endogenous mitochondria; (b) isolated exogenous mitochondria from non-senescent cells; and (c) an agent that reduces the copy number of endogenous mtDNA. In some embodiments, the agent is a fusion protein. In certain embodiments, the fusion protein comprises an mtDNA cleaving endonuclease and a Mitochondrial Targeting Sequence (MTS). In a specific embodiment, the endonuclease cleaves wild-type mtDNA. In some embodiments, the endonuclease is selected from XbaI, EcoRI, BamHI, HindIII, PstI, Cas9, Zinc Finger Nuclease (ZFN), and transcription activator-like effector nuclease (TALEN). In other embodiments, the MTS targets a mitochondrial matrix protein. In other embodiments, the mitochondrial matrix protein is selected from cytochrome c oxidase subunit IV, cytochrome c oxidase subunit VIII, and cytochrome c oxidase subunit X. In certain embodiments, the fusion protein is transiently expressed in the senescent or near-senescent cells.
The present disclosure also provides a composition for delaying senescence and/or increasing longevity in a cell, comprising: (a) senescent or near-senescent cells with endogenous mitochondria; (b) isolated exogenous mitochondria from non-senescent cells; and (c) an agent that reduces mitochondrial function. In some embodiments, the agent that decreases mitochondrial function transiently decreases endogenous mitochondrial function. In other embodiments, the agent that reduces mitochondrial function permanently reduces endogenous mitochondrial function. In some embodiments, the exogenous mitochondria from a non-senescent cell have increased function relative to the endogenous mitochondria.
In some embodiments, the composition for delaying senescence and/or extending lifespan in a cell further comprises a second active agent. In a specific embodiment, the second active agent is selected from a macromolecule, a small molecule, or a cell therapy, and the second active agent is optionally selected from rapamycin, NR (nicotinamide ribose), bezafibrate, idebenone, mercaptoethylamine bitartrate (RP103), eladimide (MTP131), omacrolone (omavelolone) (RTA408), KH176, vantiquone (Vatiquinone) (Epi743), lipoic acid, a0001 (alpha-tocopheryl quinone), mitochondrial CoQ10(MitoQ), SkQ1(Visomitin), resveratrol, curcumin, ketonic therapy, hypoxia, and an endocytic activator. In some embodiments, the endocytic activator is an activator of an endostatin-dependent endocytic pathway. In specific embodiments, the endostatin-dependent endocytosis pathway is selected from the group consisting of CLIC/GEEC endocytosis pathway, Arf 6-dependent endocytosis, lipocalin-dependent endocytosis, macropinocytosis, circular membrane ruffles (circular nuclear ruffles), phagocytosis, and trans-endocytosis. In other embodiments, the endostatin-dependent endocytic pathway is megapinocytosis. In some embodiments, the endocytic activator comprises a nutritional threat and/or an mTOR inhibitor. In certain embodiments, the mTOR inhibitor comprises rapamycin or a derivative thereof.
In another aspect, the present disclosure also provides a pharmaceutical composition comprising an isolated population of cells having mitochondrial replacement of an exogenous mitochondrion from a healthy donor, wherein the cells are obtained by any of the methods provided herein for obtaining a mitochondrial replaced cell. In another aspect, the present disclosure provides a pharmaceutical composition comprising an isolated population of cells having mitochondrial replacement of exogenous mtDNA from a healthy donor, wherein the cells are obtained by any of the methods provided herein for obtaining a mitochondrial replaced cell. In some embodiments, the pharmaceutical composition comprising an isolated population of cells having mitochondrial replacement of exogenous mtDNA from a healthy donor further comprises exogenous mitochondria.
For example, in some embodiments, a pharmaceutical composition comprising an exogenous mitochondrion from a healthy donor is obtained by a method of producing a mitochondrion-replaced cell comprising (a) contacting a recipient cell with an agent that reduces the copy number of endogenous mtDNA; (b) incubating the recipient cell with the agent for a sufficient period of time to partially reduce the endogenous mtDNA copy number in the recipient cell; and (c) co-incubating (1) the recipient cell from step (b) in which the endogenous mtDNA has been partially depleted and (2) an exogenous mitochondrion from a healthy donor for a period of time sufficient to non-invasively transfer the exogenous mitochondrion to the recipient cell, thereby producing a mitochondrion-replaced cell. In certain embodiments, the cell is obtained by a method comprising: (a) contacting the recipient cell with an agent that reduces mitochondrial function; (b) incubating the recipient cell with the agent for a period of time sufficient to partially reduce endogenous mitochondrial function in the recipient cell; and (c) co-incubating (1) the recipient cell from step (b) in which endogenous mitochondrial function has been partially reduced and (2) exogenous mitochondria from a healthy donor for a period of time sufficient to non-invasively transfer the exogenous mitochondria to the recipient cell, thereby producing a mitochondria-replaced cell.
In other embodiments, the cell is obtained by a method comprising: (a) contacting a recipient cell with an agent that reduces the copy number of endogenous mtDNA; (b) incubating the recipient cell with the agent for a sufficient period of time to partially reduce the endogenous mtDNA copy number in the recipient cell; and (c) co-incubating (1) the recipient cell from step (b) in which the endogenous mtDNA has been partially depleted and (2) the exogenous mtDNA from the healthy donor for a period of time sufficient to non-invasively transfer the exogenous mtDNA to the recipient cell, thereby producing a mitochondria-replaced cell. In other embodiments, the cell is obtained by a method comprising: (a) contacting the recipient cell with an agent that reduces mitochondrial function; (b) incubating the recipient cell with the agent for a period of time sufficient to partially reduce endogenous mitochondrial function in the recipient cell; and (c) co-incubating (1) the recipient cell from step (b) in which endogenous mitochondrial function has been partially reduced and (2) exogenous mtDNA from a healthy donor for a period of time sufficient to non-invasively transfer the exogenous mtDNA to the recipient cell, thereby producing a mitochondrial-replaced cell.
In certain embodiments of the pharmaceutical compositions provided herein, the cell is obtained by a method further comprising contacting the recipient cell with a second active agent prior to co-incubating the recipient cell with the exogenous mitochondria and/or exogenous mtDNA. In some embodiments, the second active agent is selected from a macromolecule, a small molecule, or a cell therapy, and the second active agent is optionally selected from rapamycin, NR (nicotinamide ribose), bezafibrate, idebenone, mercaptoethylamine bitartrate (RP103), elamipramide (MTP131), omaviralone (omavelolone) (RTA408), KH176, vantiquone (Vatiquinone) (Epi743), lipoic acid, a0001 (alpha-tocopherols), mitochondrial CoQ10(MitoQ), SkQ1(Visomitin), resveratrol, curcumin, a ketonic therapy, hypoxia, and an endocytic activator. In a specific embodiment, the endocytic activator is a modulator of cellular metabolism. In other embodiments, the modulator of cellular metabolism comprises nutrient starvation, a chemical inhibitor, or a small molecule. In other embodiments, the chemical inhibitor or the small molecule is an mTOR inhibitor. In other embodiments, the mTOR inhibitor comprises rapamycin or a derivative thereof.
In certain embodiments of the pharmaceutical compositions provided herein, the pharmaceutical composition further comprises a pharmaceutically acceptable carrier.
In some embodiments of the pharmaceutical compositions provided herein, the cell is a T cell. In other embodiments, the cell is a hematopoietic stem cell.
4. Description of the drawings
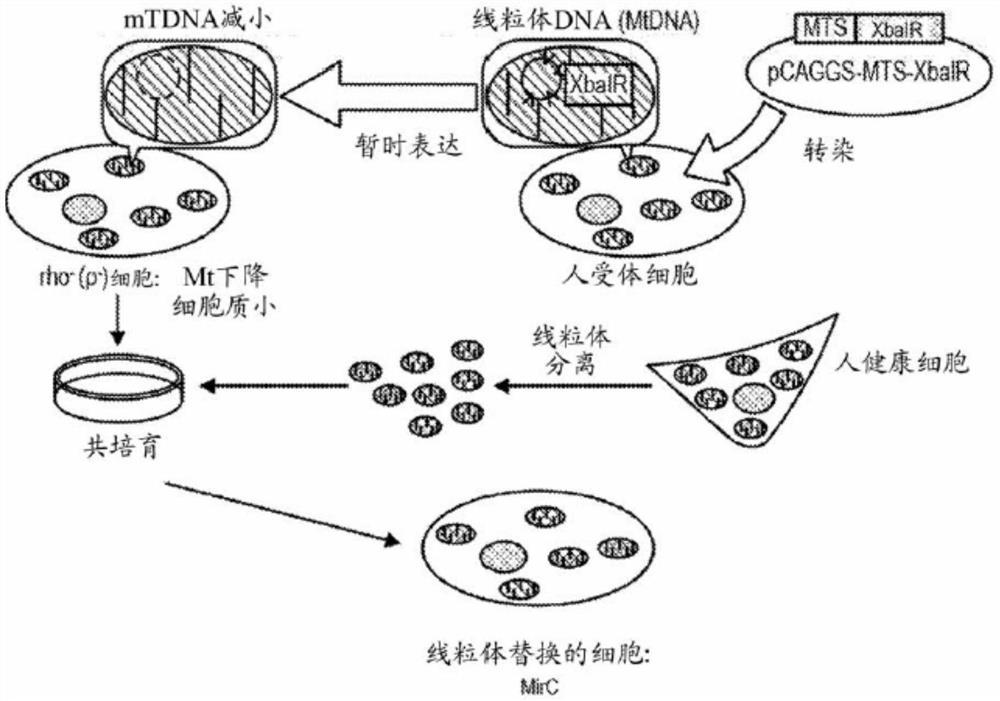
FIG. 1A shows a scheme for the production of mitochondrial-replaced cells (MirC).
FIG. 1B shows the plasmid construct of the Mitochondrial Targeting Sequence (MTS) -XbaI restriction endonuclease (XbaIR) plasmid.
Figure 1C shows isolated mitochondrial DNA digested by XbaI restriction endonuclease at multiple sites, however NotI digestion of mitochondrial DNA resulted in the generation of a single fragment as predicted by the Cambridge Reference Sequence (CRS) of mitochondrial DNA.
Figure 1D shows 5 XbaIR endonuclease sites (1193, 2953, 7440, 8286, 10256) on human mitochondrial DNA as predicted by Cambridge Reference Sequence (CRS).
Figure 1E shows the microscopy of human skin fibroblasts under phase contrast (left panel), immunofluorescence of green fluorescent protein (middle) and merged field of view (right panel) after uptake of the fused MTS-Green Fluorescent Protein (GFP) plasmid using an electrotransformation machine (Nucleofector). Top, low magnification. Bottom, high magnification.
FIG. 1F shows the constructs of pCAGGS-MTS-EGFP-Puror and pCAGGS-MTS-XbaIR-Puror plasmids.
FIG. 1G shows the localization of the exogenous transgene product MTS-EGFP in mitochondria using tetramethylrhodamine, methyl ester (TMRM) by mitochondria-specific staining.
FIG. 2A shows a schedule comparing the MTS-XbaIR endonuclease approach (top) with the conventional approach using ethidium bromide (EtBr) (middle) relative to the non-contacted cells.
FIG. 2B shows the quantification of human β -actin (Actb), left column, and mitochondrial DNA (mtDNA), right column, relative to non-contacted cells, after contact with the MTS-XbaIR endonuclease method or ethidium bromide treatment. XbaIR resulted in a greater reduction in mtDNA compared to EtBr treatment. Actb was used as a housekeeping gene.
FIG. 2C shows a greater reduction in mitochondria following gene transfer exposure to MTS-XbaIR relative to EtBr treatment based on DsRed fluorescence expressed in mitochondria.
FIG. 2D shows the semiquantitative analysis of mitochondrial membrane potential (surrogate marker for mitochondrial content) in cells contacted with gene transfer of MTS-XbaIR or EtBr using FACS analysis by using TMRM, and shows that MTS-XbaIR results in a greater reduction of mitochondria.
FIG. 2E shows the time course quantification of transgene expression within 14 days in a gene transfer system.
Fig. 2F and 2G show fluorescence images after transfer of plasmids with GFP before ("pre") and after ("post") puromycin selection (fig. 2F), and quantification of the GFP/mitochondria ratio (fig. 2G) shows enrichment of GFP plasmids after puromycin selection.
Fig. 3A shows a scheme of a schedule of mitochondrial replacement. TF: gene transfection or free-running transfection with XbaIR; puro: puromycin for gene transfer cell enrichment; u +: addition of uridine to rescue rho (-) cells lacking mitochondrial ATP production; mt Tx: mitochondrial transfer; NHDF: normal human skin fibroblasts; EPC 100: placental venous endothelium-derived cell lines.
Figure 3B shows that mitochondria decreased after gene transfer by XbaIR at day 6 (top), but not after negative control vector expression GFP transfer (bottom), as measured by TMRM staining.
Figure 3C shows quantification of mitochondrial DNA copy number estimated by qPCR of human 12S rRNA relative to nuclear β -actin levels in NHDF cells following transfection of XbaIR or GFP transfected genes. Mitochondria were transferred to the indicated recipient cells ("Mt Tx"). XbaIR results in a significant reduction of mitochondrial DNA, which can be rescued to levels comparable to control of process cells following ex vivo mitochondrial transit. N-3, p < 0.01.
Fig. 3D shows a photograph from a time interval shot (time lag movie): upper left: co-culturing rho (-) cells with isolated and DsRed-labeled mitochondria; upper right: ρ (-), as a control; left lower: co-culturing NHDF with mitochondria; right lower: co-culturing idle chromosome of NHDF with mitochondria;
FIG. 3E shows a series of 10 still images taken from the time interval shown in FIG. 3D arranged vertically in a time sequence;
FIG. 3F shows measurement of DsRed-tagged mitochondria by FACS analysis and shows that the present invention ("DsRed-Mt EPC 100") results in increased exogenous mitochondrial uptake compared to previously described methods.
Figures 3G and 3H show microscopical pictures of DsRed-labeled mitochondria (figure 3G) and phase contrast (figure 3H) after mitochondrial transfer in ρ (0) cells with or without antimycin treatment, and indicate that no exogenous mitochondrial engulfment occurred in fully mitochondria-disrupted cells.
Fig. 3I shows a series of 5 still images from the time interval shot shown in fig. 3G arranged vertically in time sequence.
FIG. 3J shows the quantification of fluorescence intensity of DsRed-labeled isolated exogenous mitochondria measured every 24 hours in either rho (-) cells or rho (-) idle stained cells co-incubated with Ds-red mitochondria or untreated cells (additional Mt).
Figure 4A shows a protocol for measuring the fate of donor mitochondria after engulfment by recipient cells using DsRed-labeled mitochondria as donor and EGFP-labeled cells as recipient.
Fig. 4B shows representative images taken from observations of engulfed foreign mitochondria (indicated as red) in recipient cells with GFP-labeled mitochondria. The shots were recorded by using an ultra-precision microscope and a few fusion images were identified and most of the donor mitochondria were present alone in pre-existing mitochondria.
Fig. 4C shows a photograph of the fused stereo reconstruction.
FIG. 4D shows photographs of NHDF transfer of DsRed encoding gene fused to mitochondrial transfer signal.
Figure 4E shows photographs of EPC100 transfer encoding EGFP fused to TFAM.
Fig. 4F shows the time course of mitochondrial transfer using DsRed labeled cells as recipients and TFAM targeted EGFP as the donor mitochondria.
Figure 4G shows that exogenous TFAM was stably implanted into pre-existing mitochondria after transient contact of the exogenous mitochondria with recipient cells, suggesting that mitochondrial pseudokaryons including TFAM were transferred into pre-existing mitochondria by transient contact, similar to mouth-to-mouth uptake.
FIG. 5A shows complete circular mitochondrial DNA with Cambridge Reference Sequence (CRS) indicating the high variation ("HV") region 1/2 and 5 primers to identify human mitochondrial DNA for differences between NHDF and EPC 100;
FIG. 5B shows DNA sequencing data for nucleotides around hmt16362 in NHDF control recipient cell (SEQ ID NO: 1), EPC100 control donor cell (SEQ ID NO: 2), NHDF derived rho (-) cell with NO mitochondrial replacement (SEQ ID NO: 3) and NHDF derived rho (-) cell with mitochondrial replacement (SEQ ID NO: 4) and shows that rho (-) cell with mitochondrial replacement (SEQ ID NO: 4) changed from A in the original recipient cell to G in the donor mtDNA at hmt 16362.
FIG. 5C shows primer sets of hmt16318-F (SEQ ID NO: 6) and hmt16414-R (SEQ ID NO: 9) for amplification of the HV1 region (SEQ ID NO: 8) of the D-loop of human mitochondrial DNA surrounding hmt16362, as well as NHDF specific probe (SEQ ID NO: 5) and EPC100 specific probe (SEQ ID NO: 7) designed for TaqMan SNP genotyping assays.
Fig. 5D shows quantification of NHDF-specific hmtd (left panel) and EPC 100-specific hmtd (right panel) in parental NHDF and EPC100 cell lines, or NHDF cells treated with XbaIR, with (XbaIR Mt +) or without mitochondria from EPC100 cells (XbaIR Mt-), and shows successful transfer of EPC100 mitochondria into XbaIR Mt + cells, as assessed by using single nucleotide polymorphism assay (SNP).
Fig. 6A shows a representative aerograph (Oxygraph) from mitochondrial function assays performed using orobos Oxygraph-2k and shows that NHDF cells with mitochondrial replacement (ρ (-) Mt) (bottom) restored mitochondrial function relative to control NHDF cells (top) and wirelessly mitochondrial replaced ρ (-) NHDF cells (middle). The machine shows the respiratory flow (pmol/sec/1X 10) in red6Individual cells, right axis) and the oxygen concentration (μ M, left axis) is shown in blue line.
Fig. 6B shows respiratory flow (conventional, Electron Transfer System (ETS), ROX), free conventional activity (mitochondrial ATP production), proton leak, and coupling efficiency at each stage indicating that mitochondrial function was restored by mitochondrial replacement in NHDF cells (ρ (-) Mt) relative to NHDF control cells and NHDF (ρ (-)) without mtDNA replacement.
Fig. 6C shows time interval micrographs that enable the estimation of the number of consecutive cells based on cell surface area and demonstrate that ρ (-) cells are quiescent between days 3 and 12, whereas mitochondria-replaced cells recovered growth capacity after day 6.
FIG. 6D shows a protocol for examining the molecular mechanisms of megacaryon, which involves transfecting NHDF cells with the MTS-XbaIR-P2A-Puror plasmid, selecting with puromycin, and then serum starving the cells for 60min or treating the cells with Palmitic Acid (PA) or rapamycin for 24 hours.
FIGS. 6E-6H show WES for kinase phosphorylation of S6 (FIG. 6E) and AMPK phosphorylation (FIG. 6G)TMAnalysis, and respectively corresponding WESTMQuantification of blots (fig. 6F) and (fig. 6H) indicating that AMPK is activated and mTOR is completely inhibited in ρ (-) cells. Rapa: rapamycin, PA: palmitic acid, EAA-: essential amino acid-deficiency.
Figure 6I shows the protocol used to examine the effect of mTOR-mediated megapinocytosis in the context of the MirC production protocol.
Fig. 6J-6L show quantification of DsRed labeled mitochondrial uptake (fig. 6J and 6K) and FACS analysis (fig. 6L) in control (top), free-stained cells (middle) and ρ (-) cells treated with or without rapamycin or with or without Palmitic Acid (PA). Rho (-) cells showed greater mitochondrial uptake relative to the empty TF cell control, and mitochondrial uptake was significantly increased after rapamycin treatment, whereas palmitic acid reduced mitochondrial uptake in rho (-) cells.
Figure 7A shows the complete mtDNA sequence showing the Leigh syndrome-associated mutation of 10158T > C in the respiratory chain complex i (ci) subunit of the ND3 gene in mitochondrial DNA.
FIG. 7B shows DNA sequencing data for nucleotides surrounding hmt10158 within ND3 in EPC100 cells (top; SEQ ID NO: 10) and Leigh syndrome (7SP) fibroblasts (bottom; SEQ ID NO: 11), and shows the mutation 10158T > C as a mosaic of C in the dominant wave and T in the subordinate wave, thus indicating heterogeneity.
Fig. 7C shows photographs taken from time intervals, which indicate that the situation is similar in both ρ (-)7SP fibroblasts with and without exogenous mitochondria, as in the NHDF experiment.
Figure 7D shows quantification of mitochondrial DNA copy number estimated by qPCR of human 12S rRNA, relative to nuclear β -actin levels in NHDF cells, after XbaIR gene transfection or free-running staining. Mitochondria were transferred to the indicated recipient cells. XbaIR results in a significant reduction of mitochondrial DNA, which can be rescued by exogenous mitochondrial transfer. (n is 3)
FIG. 7E shows DNA sequencing data for nucleotides surrounding hmt10158 in 7SP control recipient cells (SEQ ID NO: 14), EPC100 control donor cells (SEQ ID NO: 12), 7 SP-derived rho (-) cells without mitochondrial replacement (SEQ ID NO: 13), and 7 SP-derived rho (-) cells with mitochondrial replacement (SEQ ID NO: 15), and indicates that the 7SP control cells are heterogeneous (mostly 10158C; SEQ ID NO: 14), whereas EPC100 has a T (SEQ ID NO: 12) only at the same site in mitochondrial DNA. Rho (-) cells derived from 7SP cells expressed the same wave as the original (SEQ ID NO: 13), whereas mitochondria-replaced 7SP cells showed T as the dominant wave (SEQ ID NO: 15).
FIG. 7F shows the hmt10085-F (SEQ ID NO: 17) and hmt10184-R (SEQ ID NO: 20) primer sets for amplification of ND3(SEQ ID NO: 16) of human mitochondrial DNA surrounding the Leigh syndrome associated SNP at hmt10158, as well as the EPC 100-specific probe (SEQ ID NO: 18) and the 7 SP-specific probe (SEQ ID NO: 19) designed for TaqMan SNP genotyping assays. The peptide sequence ND3 is also shown (SEQ ID NO: 46).
Fig. 7G shows quantification of the percentage of hmt10158 heterogeneity in each cell group assessed by SNP assay and shows predominance of exogenous normal sequences ("healthy") and up to 80% in mitochondria-replaced 7SP cells, even though the original heterogeneity of mutant sequences was over 90%. In the case of the idle dye, the heterogeneity did not change significantly and maintained nearly the same ratio.
Fig. 7H and 7I show quantification of the percent level of heterogeneity (fig. 7H) and absolute mtDNA copy number (fig. 7I) in 7SP cells treated with empty controls and subjected to mitochondrial transfer in 3 independent experiments.
Fig. 7J shows a series of 10 still images from the time interval shooting shown in fig. 7G arranged vertically in time series.
Figure 8A shows micrographs of ρ (-) mitochondrial displaced 7SP fibroblasts over time compared to the original 7SP fibroblasts and ρ (-)7SP fibroblasts and shows that the growth of the mitochondrial displaced cells was restored to near control levels.
Fig. 8B shows the cell growth estimated over time in 7SP fibroblasts, ρ (-)7SP fibroblasts, and ρ (-)7SP fibroblasts with mitochondrial replacement, whereas the mitochondrial replacement 7SP cells restored cell growth to levels comparable to the original 7SP fibroblasts at about day 12.
Fig. 8C shows senescence in 7SP fibroblasts at Population Doubling Level (PDL) of about 25, which was prolonged to about PDL 63 in ρ (-)7SP fibroblasts with healthy mitochondrial replacement performed with PDL 8, indicating a prolonged lifespan of ρ (-)7SP fibroblasts with healthy mitochondrial replacement.
Fig. 8D shows that an increase in PDL produces an increase in cell size (left panel), which recovers after mitochondrial replacement and remains even beyond PDL 50 (right panel).
Figure 8E shows a Short Tandem Repeat (STR) assay that distinguishes cells with different origins and identifies contamination of different types of cells. At different time points, the STR types in the mitochondria-replaced cells were identical to those in the original 7SP fibroblasts.
Fig. 8F shows RT-PCR quantification of telomerase in 7SP fibroblasts and mitochondria-replaced cells for different PDL, relative to HeLa and EPC100, indicating that the cells were not transformed into cancer cells.
Fig. 9A shows the oxygen tracings in different PDL, 7SP fibroblasts after mitochondrial replacement with orobos O2k according to the coupling-control protocol (CCP), and the kinetics indicate that mitochondrial function is reduced in early PDL, then slowly restored and eventually exceeds the original capacity, relative to the original 7SP fibroblasts as a control.
Fig. 9B and 9C show that after about PDL30, respiratory flux (conventional, Electron Transport System (ETS), ROX), free conventional activity (mitochondrial ATP production), proton leakage and coupling efficiency (fig. 9B), and Flux Control Ratio (FCR), ROX/E, L/E, R/E, and (R-L)/E (fig. 9C) were restored to near control levels in mitochondria-replaced cells (ρ (-) Mt).
FIG. 10A shows the results under basal conditions or in the use of H2O2After reperfusion, microscopic images of NHDF, 7SP MirC cells and showing 7SP cell pairs H relative to NHDF cells2O2Highly sensitive, however 7SP MirC is not.
FIGS. 10B-10D show the treatment without treatment or with H2O2FACS analysis (FIG. 10B) and quantification of annexin V (FIG. 10C) and propidium iodide (PI; FIG. 10D) positive cells after treatment and showed 7SP cells vs. NHDF cells for H 2O2Highly sensitive, however 7SP MirC is not.
Fig. 10E shows microscopic images of NHDF, 7SP MirC cells under basal conditions or after starvation conditions (EAA-) and shows that 7SP cells are highly sensitive to starvation conditions relative to NHDF cells, whereas 7SP MirC is not.
Fig. 10F-fig. 10H show FACS analysis (fig. 10F) and quantification of annexin V (fig. 10G) and PI (fig. 10H) positive cells after no treatment or starvation treatment, and show that 7SP cells are highly sensitive to starvation conditions relative to NHDF cells, whereas 7SP MirC is not.
Figure 11 shows quantification of expression levels of representative SASP cytokines IL-6 and IL-8, chemokine CXCL-1 and growth factor ICAM1 for NHDF, 7SP fibroblasts and 7SP fibroblast-derived MirC cells with nearly identical PDL (about 15 to 20), indicating a significant decrease in IL-6, indicating reversal of SASP in MirC. GAPDH was used for normalization.
Fig. 12A shows a generation scheme of induced pluripotent stem cells (ipscs) from mitochondria-replaced 7SP fibroblasts.
Fig. 12B-12D show Alkaline Phosphatase (AP) staining and quantification as an indicator of ipscs, generated from 7SP fibroblasts, 7SP fibroblast-derived MirC, or an idling stain derived from 7SP fibroblasts. Microscopic (fig. 12B left panel; 7SP fibroblasts, middle panel; 7SP fibroblast-derived MirC, right panel: 7SP fibroblast idle chromosome) and macroscopic (fig. 12C left panel; 7SP fibroblasts, middle panel; 7SP fibroblast-derived MirC, right panel: 7SP fibroblast idle chromosome) microscopy and quantification of AP stained cells (fig. 12D) showed that mitochondrial replacement in NHDF or 7SP fibroblasts resulted in increased AP staining after XbaIR treatment.
Figure 12E shows colony formation of ipscs derived from mitochondrially replaced 7SP fibroblasts. Photographs of 3 representative colonies at 75 days and 170 days after gene transfer of the reprogramming factors.
FIG. 12F shows immunohistochemical staining of OCT3/4, NANOG, TRA1-80, and TRA-160 in iPSC generated from 7SP fibroblasts following mitochondrial replacement, which is a representative marker for pluripotent stem cells;
fig. 12G shows the mitochondrial DNA copy number in ipscs derived from 7SP fibroblast-derived MirC compared to original 7SP fibroblasts and standard human ipscs as reference (201B7), and shows that ipscs have a similar limited mitochondrial DNA number as standard human ipscs.
Fig. 12H and 12I show the percentage of heterogeneity (fig. 12H) and absolute mtDNA copy number (fig. 12I) in ipscs derived from 7SP fibroblast-derived MirC 170 days after reprogramming, and show that 7SP fibroblast-derived MirC forming ipscs showed negligible levels of mutant genomic sequence, reduced total mtDNA and close to 100% donor mtDNA in at least 3 clones, suggesting that the heterogeneity change in MirC can revert to the original state and is different from mitochondrial replacement therapy in IVF.
Figure 13A shows a protocol for mitochondrial transfer from a donor cell to a recipient cell, where the donor cell and recipient cell are from different stages of the life cycle.
FIG. 13B shows DNA sequencing data for nucleotides surrounding hmt16145 in NHDF control recipient cells (SEQ ID NO: 21) with genotype hmt 16145A and TIG1 control donor cells (SEQ ID NO: 22) with genotype hmt 16145G.
Fig. 13C shows quantitation of hmt16145 heterogeneity levels (%) by SNP assay of cells from mitochondria-replaced cells (mircs) ("old" NHDF recipient cells with mitochondrial transfer from mitochondria of "young" TIG1 donor cells) and indicates that greater than 90% of mtDNA in NHDF-derived MirC cells with mitochondrial replacement from TIG 1-derived mitochondrial donor cells is hmt 16145G (i.e., from TIG1 mtDNA), whereas 100% of the mtDNA of NHDF control cells is hmt 16145A.
Fig. 13D shows quantification of Population Doubling Levels (PDL) versus time (days) (left panel) and doubling times (hours) versus population doubling levels (right panel) in recipient NHDF cells transfected with MTS-GFP ("null") or MTS-XbaIR ("MirC") and co-incubated with exogenous mitochondria from TIG1 donor cells or untransfected recipient NHDF cells ("control"). MirC with replacement of "young" donor TIG1 embryonic lung cells (PDL 10) versus "old" Normal Human Dermal Fibroblast (NHDF) recipient cells (PDL 41) showed an extended lifespan as shown by an upward shift in PDL (left panel) and a rightward shift in PDL (right panel).
Figure 13E shows quantification of Population Doubling Levels (PDL) versus time (days) (left panel) and doubling times (hours) versus population doubling levels (right panel) in normal human dermal fibroblasts transfected with MTS-GFP plus mitochondrial transfer ("null") or MTS-XbaIR plus mitochondrial transfer ("MirC") or untransfected normal human dermal fibroblasts ("control"). Mitochondrial transfer from "old" donor cells (PDL 49) to "young" recipient cells (PDL <21) showed a shortened lifespan as shown by downward migration in PDL (left panel) and leftward migration in PFL (right panel).
FIG. 14A shows an assessment of the quality of mRNA produced by in vitro transcription as measured by electrophoresis of mRNA for MTS-EGFP and MTS-XbaIR.
FIG. 14B shows strong expression of the MTS-GFP transgene in mitochondria of T cells 24 hours after electroporation.
Fig. 14C shows FACS analysis of GFP expression in T cells after transfection of MTS-GFP mRNA by electroporation and shows that GFP expression is present in nearly all T cells.
Fig. 14D shows FACS analysis of DsRed labeled mitochondria and demonstrates that the MTS-XbaIR construct robustly degrades endogenous mitochondria, whereas MTS-GFP does not.
Fig. 14E shows the protocol design for determining the optimal time period for mitochondrial co-incubation.
Fig. 14F shows fluorescence images of control electroporated cells (upper panel) and MTS-GFP electroporated cells (lower panel) at 4 hours, 2 days, 4 days, 6 days, and 8 days after Electroporation (EP), and indicates that within 4 hours after electroporation, the MTS-GFP construct showed high expression and was almost absent by day 6.
FIGS. 14G and 14H show the electrophoresis (FIG. 14H) and quantification (FIG. 14G) of GFP in cells receiving MTS-GFP mRNA relative to GAPDH. Peak expression occurred on day 4, and by day 6, expression disappeared.
FIG. 14I shows quantification of XbaIR transcript levels at 4 hours, day 2 (d2), day 4 (d4), day 6 (d6) and day 8 (d8), indicating that endonuclease transcript expression was highest 4 hours after gene transfer.
Figure 14J shows quantification of mitochondrial content in cells subjected to MTS-XbaI (12S rRNA) and shows that by day 2, mitochondria decreased to about 30% and remained at less than 20% throughout the length of the experiment.
Fig. 15A shows the MirC protocol for human primary T cells electroporated at day 0, analyzed at day 2, mitochondrial (mt) transfer at day 7, SNP assay at days 9 and 14, and ddPCR heterogeneity assay at day 14.
FIG. 15B shows DNA sequencing data for nucleotides around hmtDNA 218 and hmtDNA 224 of the HV1 region of the human mitochondrial DNA D-loop in human primary NH T cell control recipient cells (top; SEQ ID NO: 23) and EPC100 control donor cells (bottom; SEQ ID NO: 24). For T cells and EPC100 cells, hmtDNA 218 and hmtDNA 224 are C/C (SEQ ID NO: 23) and T/T (SEQ ID NO: 24), respectively.
FIG. 15C shows Primer sets for amplification of hmtHV1-F (SEQ ID NO: 26) and hmtHV1-R (SEQ ID NO: 27) for the HV1 region of human mitochondrial DNA D-loop (SEQ ID NO: 25) surrounding SNPs at hmtDNA 218 and hmtDNA 224, as well as SNP assay primers Primer1-F (SEQ ID NO: 40), SNP assay Primer1-R (SEQ ID NO: 41), an N-terminal VIC-labeled EPC 100-specific probe (SEQ ID NO: 38) and an N-terminal FAM-labeled T-cell-specific probe (SEQ ID NO: 39) designed for TaqMan SNP genotyping assays.
FIG. 15D shows quantification of the amount of exogenous mtDNA present in recipient cells on days 7 and 12 for null (MTS-GFP) or MTS-XbaIR (XbaIR) treated cells after co-incubation with exogenous mitochondria from donor EPC100 cells. Quantification of recipient and donor cells was performed as a positive control.
Fig. 15E shows quantification of the respiratory measurement experiment using orobos O2k and demonstrates recovery of ATP production and coupling efficiency in human T cell-derived MirC, whereas ρ (-) human T cells generated by transfer of XbaIR mRNA using electroporation maintained loss of ATP production throughout the experiment.
Fig. 15F and 15G show representative raw data using the coupling-control protocol (CCP) and show that MirC T cells are able to restore mitochondrial respiration.
Fig. 16A shows a comparison of viability (left panel) or CD3 expression (right panel) of mouse primary T cells cultured in RPMI1640 (top) or TexMACS (bottom) at day 2 (left panel left), day 4 (middle left panel) and day 6 (right panel left), and shows that RPMI1640 produced greater viability and higher cell counts, and a slight increase in CD3 expression relative to TexMACS medium.
FIG. 16B shows a qualitative analysis of GFP expression in T cells following Electroporation (EP), using pmax GFP (middle) or MTS-GFP (right panel) or without electroporation (left panel), at 6 hours post-EP (top left panel), 2 days post-EP (top right panel), 4 days post-EP (bottom left panel) and 6 days post-EP (bottom right panel). The use of MTS-GFP did not significantly affect viability at either 2 or 4 days post EP.
Figure 16C shows qPCR quantification of XbaIR levels in electroporated T cells using MTS-XbaIR vector at 4 hours, 2 days, 4 days, and 6 days post-electroporation and indicates a slow decrease in XbaIR expression.
Figure 16D shows quantification of 12S rRNA levels in electroporated T cells using MTS-XbaIR and indicates that murine mtDNA decreased by about 60% by day 4.
Figure 16E shows the protocol for MirC production in T cells on day 5 using mitochondrial co-incubation.
FIG. 16F shows FACS analysis of DsRed-labeled mitochondria engulfed in recipient T cells 48 hours after co-incubation with isolated DsRed-labeled mitochondria, and shows a significant positive fraction (9.73%) of T cells expressing outer mitochondria in MTS-XbaIR compared to 0.43% in control cells without electroporation (i.e., "spiked").
FIG. 17A shows DNA sequencing data for nucleotides surrounding ND1 in mouse mtDNA C57BL6 recipient cells ("BL 6"; top; SEQ ID NO: 34) with genotypes mmt2766-A and mmt2767-T and NZB donor cells (bottom; SEQ ID NO: 35) with genotypes mmt2766-G and mmt 2767-C.
FIG. 17B shows the primer set of 2716-F (SEQ ID NO: 28) and 2883-R (SEQ ID NO: 33) for amplification of ND1(SEQ ID NO: 32) of mouse mitochondrial DNA surrounding the polymorphic nucleotides mmt2766 and mmt2767, and BL 6-specific probe (SEQ ID NO: 29) and NZB-specific probe (SEQ ID NO: 31) designed for TaqMan SNP genotyping, as well as BamH1-mND1-F primer (SEQ ID NO: 30) for cloning of nucleotide sequences in plasmids used to generate standard curves capable of absolute quantification. The peptide sequence ND1 is also shown (SEQ ID NO: 47).
Figure 17C shows quantification of mouse mtND1 heterogeneity levels in BL6 receptor cells 7 days and 12 days after control electroporation (columns 1 and 2, respectively) or MTS-XbaI electroporation and co-incubation with isolated mitochondria from NZB cells (columns 3 and 4, respectively). Basal levels of BL6 (column 5) and NZB (column 6) cells were measured as controls.
FIG. 17D shows measurement of telomere length after treatment of old murine cells with MTS-XbaIR mRNA and co-incubation with exogenous mitochondria from young donor cells to produce MirC (young to old: YtoO) and indicates increased telomere length in MirC compared to parental "old" cells.
FIG. 17E shows the measurement of the SASP-associated cytokines CXCL1, ICAM1, IL-6 and IL-8 in parental old T cells or MirC-derived T cells and indicates that CXCL1 and IL6 are less in MirC-derived T cells.
Fig. 17F shows measurement of DNA damage response in MirC and naive T cells using histone 2A (H2A) phosphorylated antibody, which indicates a lower DDR positive fraction (1.53%) in MirC compared to naive T cells (4.75%).
Fig. 18A shows an in vivo ACT experimental protocol using old mice with ACT from T cells from young mice (group 1), old mice with ACT (group 2), or old mice with ACT derived from MirC transferred T cells from old mice of exogenous mitochondria from young mice (group 3).
Fig. 18B shows representative images of tumor growth imaging performed during the experimental protocol.
Figure 18C shows body weights of empty, young T cells or MirC groups and indicates that no significant difference was observed between the 3 groups during the 25 day experiment.
Figures 18D and 18E show the quantification of individual (figure 18D) and average (figure 18E) cancer mass sizes and indicate that the MirC group reduced the cancer mass size to levels equivalent to the young T cell group (lower line), whereas the cancer mass in the empty group increased during the entire length of the experiment (upper line).
Figure 18F shows a protocol for analyzing animals for the presence of infused T cells.
Fig. 18G shows FACS analysis of peripheral blood (left panel) or spleen (right panel). Negative controls using C57BL/6 mice (top left panel) and positive controls using GFP transgenic mice (bottom left panel) were generated for both peripheral blood and spleen. Positive fractions of GFP-fluorescent expressing T cells were identified in both peripheral blood and spleen, which were 0.057% and 0.9%, respectively.
FIG. 18H shows immunofluorescence images of metastatic T cells detected in mice at day 6 post-transplantation.
FIG. 18I shows the implant at 1X 107Or 2X 107Percentage chimerism after infusion of exogenous T cells in Peripheral Blood (PB) or spleen after one cell.
FIGS. 19A and 19B show the evaluation of MTS-GFP transfection into hematopoietic cells (HSCs) by microscopy (FIG. 19A) or FACS (FIG. 19B) using either the X-001, Y-001 and T-030 programs (MTS-GFP 1, 2 and 3, respectively) or pmax GFP as a positive control or Ctl EP as a negative control, and MTS-GFP1 is the optimal protocol for electroporating HSCs.
FIG. 19C shows three-dimensional confocal fluorescence imaging of bone marrow-derived Sca-1 cells 48 hours after co-incubation with DsRed-labeled mitochondria from EPC100 cells, and shows engulfment of exogenous mitochondria.
FIG. 19D shows quantification of mitochondrial transfer efficiency by FACS analysis of DsRed fluorescence and shows that about 10% of the Sca-1 subpopulation shows rightward migration of fluorescence.
Fig. 19E shows a protocol for generating HSC-derived mircs by co-incubation with exogenous mitochondria at day 4 and analysis of mircs by SNP assay at day 6.
FIG. 19F shows FACS sorting of the c-kit +, Sca-1+, pedigree-, CD34- (referred to as KSLC) portion of the cells.
FIG. 19G shows that the doubling time for the KSLC fraction is 19 hours.
Fig. 19H shows a protocol for evaluating HSC-derived MirC.
FIG. 19I shows quantification of the percentage of murine mtND1 heterogeneity levels in murine KSLC-derived MirC or parental recipient BL6 cells or NZB donor cells and indicates that MirC-derived HSCs express a polymorphic genotype of 99.9% of the donor cells 6 days after MTS-XbaI mRNA transfer by electroporation.
Fig. 20A shows a 2-D plot of the droplet digital PCR results for tRNA Leu 3243A > G in which the mutated mtDNA and non-mutated mtDNA sequences were analyzed in normal human dermal fibroblasts, and only shows that non-mutated sequences were detected (lower right quadrant) and no mutated sequences were detected (upper left quadrant).
Fig. 20B shows a 2-D plot of the microdroplet digital PCR results of ND 310158T > C in which mutated and non-mutated mtDNA sequences were analyzed in normal human dermal fibroblasts, and only shows that non-mutated sequences were detected (lower right quadrant) and no mutated sequences were detected (upper left quadrant).
Fig. 20C shows a 2-D plot of the droplet digital PCR results for ATP 69185T > C in which mutated mtDNA and non-mutated mtDNA sequences were analyzed in normal human skin fibroblasts, and only shows that non-mutated sequences were detected (lower right quadrant) and no mutated sequences were detected (upper left quadrant).
Fig. 20D shows a 2-D plot of the droplet digital PCR results where mutant and non-mutant mtDNA sequences were analyzed in primary dermal fibroblasts from patients with MELAS having the mtDNA a3243G mutation, and shows that most cells have homoheterogeneity of mutant mtDNA (upper left quadrant).
Figure 20E shows a 2-D plot in which the microdroplet digital PCR results of mutant mtDNA and non-mutant mtDNA sequences were analyzed in primary dermal fibroblasts from patients with Leigh syndrome with the mtDNA T10158C mutation of the complex I, ND3 gene, and double positive cells with a minor portion of heterogeneity at the single cell level (upper right quadrant), and showing that most of the population had homoheterogeneity of mutant mtDNA (lower right) and none of the population had homoheterogeneity of non-mutant mtDNA (upper left).
5.Detailed description of the invention
Provided herein are novel and enhanced methods of producing mitochondrial-replaced cells (MirC) that do not require complete removal of endogenous mtDNA and that can optionally be performed using reagents compatible with clinical use. Additionally, in certain embodiments, provided herein are methods of treatment comprising administering a therapeutically effective amount of MirC produced using the methods provided herein.
Also provided are compositions comprising one or more mitochondria-replaced cells obtained by the methods provided herein. In certain embodiments, the composition can further comprise a second active agent that increases uptake of exogenous mitochondria, exogenous mtDNA, or a combination thereof and/or an agent that reduces the copy number of endogenous mtDNA or an agent that reduces endogenous mitochondrial function. In other embodiments, the composition may further comprise exogenous mitochondrial and/or exogenous mtDNA, one or more recipient cells, or a combination thereof. In a specific embodiment, provided herein are methods and compositions for use in treating a disease or disorder associated with dysfunctional mitochondria. However, it is to be understood that the methods and compositions provided herein may also be used to delay senescence, extend the lifespan or enhance the function of cells with functional mitochondria, and are not limited to the replacement of dysfunctional mitochondria. In addition, the methods and compositions provided herein can also be used to replace functional mitochondria (for example) with dysfunctional or depleted exogenous mitochondria to generate disease models.
5.1Definition of
Unless specifically defined otherwise, all terms, including technical and scientific terms, used in the present application have the same meaning as commonly understood by one of ordinary skill in the art to which this invention belongs. Generally, nomenclature used in this specification and the experimental methods described below are those commonly known and used in the relevant art.
As used herein, the term "mitochondrial-substituted cell" or MirC is intended to mean a cell with replacement of exogenous mitochondrial and/or mtDNA for endogenous mitochondrial and/or mtDNA. For example, an exemplary mitochondrial-replaced cell (MirC) comprises replacing endogenous mtDNA encoding dysfunctional mitochondria, such as mtDNA from a subject suffering from a mitochondrial disease or disorder, with exogenous mtDNA encoding functional mitochondria, such as mtDNA from a healthy subject. Exemplary mircs can also comprise cells that replace endogenous mitochondria with exogenous mitochondria. However, it is to be understood that replacement of endogenous mitochondrial and/or mtDNA may also include, for example, replacement of functional endogenous mtDNA from one cell, such as from an aged cell, with functional exogenous mtDNA from a different cell, such as a healthier cell from a young subject. It will also be appreciated that healthy endogenous mitochondrial and/or mtDNA may also be replaced with dysfunctional exogenous mitochondrial and/or exogenous mtDNA, such as, for example, to mimic a mitochondrial disease or disorder. Substitutions need not result in complete replacement of all endogenous mitochondria in a cell, and exemplary mitochondrial and/or mtDNA substitutions include substitutions of about 5% or more, about 10% or more, about 20% or more, about 30% or more, about 40% or more, about 50% or more, about 60% or more, about 70% or more, about 80% or more, about 90% or more, or about 95% or more of endogenous mitochondria and/or mtDNA.
As used herein, the terms "recipient cell", "recipient cell" and "host cell" are interchangeable and refer to a cell that receives exogenous mitochondrial and/or mtDNA. In some embodiments, the exogenous mitochondrial and/or mtDNA is from an isolated mitochondrion from a donor cell. In some embodiments, the donor cell and the recipient cell may be different or the same. In some embodiments, the donor cell and the recipient cell are from different or the same species. In some embodiments, the donor cell and the recipient cell are from different or the same tissue.
As used herein, the term "healthy donor" is intended to mean a donor of mitochondria that does not suffer from a mitochondrial disease or disorder, an age-related disease, or other dysfunction. In a preferred embodiment, a healthy donor has a wild-type mtDNA sequence relative to the cambridge reference sequence of the mitochondrial genome.
As used herein, the term "treatment" means a reduction in the severity, progression, spread and/or frequency of symptoms, elimination of symptoms and/or underlying causes, prevention of the occurrence of symptoms and/or their underlying causes, and amelioration or remediation of injury. "treatment" is meant to include both therapeutic treatment of a condition, disease or disorder as well as prophylactic or inhibitory measures.
As used herein, the term "agent" when used in reference to eliminating, reducing mtDNA, refers to an enzyme or compound capable of reducing mtDNA. Preferred agents include restriction enzymes, such as XbaI, which cleave mtDNA at one or more sites without generating toxicity in the recipient cell. However, the agent may also include an enzyme or compound that inhibits mtDNA synthesis or selectively promotes mitochondrial degradation.
As used herein, the term "reduce" or "reduction" generally refers to a reduction of at least 5%, e.g., at least about 10%, or at least about 20%, or at least about 30%, or at least about 40%, or at least about 50%, or at least about 60%, or at least about 70%, or at least about 80%, or at least about 90%, or any reduction between 5% and 99%, as that term is defined herein, as compared to a reference level. As used herein, it is understood that an agent that partially reduces or partially reduces endogenous mtDNA or a reduction does not result in complete elimination of all endogenous mtDNA (i.e., ρ 0 cells). The term "increase" as used herein generally means an increase of at least 5%, for example, an increase of at least about 10%, or at least about 20%, or at least about 30%, or at least about 40%, or at least about 50%, or at least about 60%, or at least about 70%, or at least about 80%, or at least about 90%, or greater than 90%.
As used herein, the term "endogenous" refers to being produced or derived internally. For example, an endogenous mitochondrion is a mitochondrion that is native to the cell.
As used herein, the term "exogenous" refers to cellular material that is non-native to the host (e.g., mitochondria or mtDNA), such as cellular material of external origin. "external" generally means from a different source. For example, a mitochondrial genome is foreign to a host cell or host mitochondria when it is derived from a different cell type or a different species than the host cell or host mitochondria. In addition, "exogenous" may also refer to the mitochondrial genome that is removed from mitochondria, manipulated, and returned to the same mitochondria.
As used herein, the term "sufficient period of time" refers to the amount of time to produce the desired result. It is understood that the sufficient period of time will vary depending on the experimental conditions, including but not limited to, temperature, amount of reagent used, and cell type. As a guide, an exemplary protocol is provided herein throughout for "a sufficient period of time" and one skilled in the art will be able to identify a sufficient period of time without undue experimentation.
As used herein, the term "majority" is intended to mean the largest amount relative to another amount compared. When comparing two groups, an exemplary majority is an amount of any integer greater than about 50% or more, about 60% or more, about 70% or more, about 80% or more, or about 90% or more, or about 95% or more of the total population, including any integer therebetween. It will be understood that most will depend on the total population compared and may be an amount of less than 50% when there are 3 or more groups compared.
As used herein, the term "non-invasive" when used to refer to the transfer of exogenous material is intended to mean that no invasive instruments (e.g., nanopipettes or electroporation), physical forces (e.g., centrifugation), or deleterious culture conditions (e.g., thermal shock) are used. In a preferred embodiment, the non-invasive transfer procedure comprises co-incubation of recipient cells and donor mitochondria.
As used herein, the term "subject in need of mitochondrial replacement" is intended to mean a subject that has or is predisposed to having dysfunctional mitochondria. Subjects in need of mitochondrial replacement may be asymptomatic and in need of prophylactic care. Subjects in need of mitochondrial replacement may also be symptomatic and in need of treatment. In certain embodiments, a subject in need of mitochondrial replacement has mitochondria that are not dysfunctional due to an age-related disease or a mitochondrial disease or disorder.
As used herein, the term "subject" is intended to mean a mammal. The subject may be a human or non-human mammal, such as a dog, cat, cow, horse, mouse, rat, rabbit or transgenic species thereof. It is to be understood that "subject" may also mean a "patient", such as a human patient.
As used herein, the term "effective amount" refers to an amount of a composition of the invention effective to modulate, treat, or ameliorate any disease or condition associated with heterogeneous and/or dysfunctional mitochondria. As such, an effective amount may include, for example, a therapeutically effective amount, which refers to a therapeutically effective amount or a biologically effective amount, which refers to an amount effective for a biological effect. The terms "therapeutically effective amount" and "effective amount" can encompass an amount that improves overall therapy, reduces or avoids symptoms or causes of a disease or disorder, or increases the therapeutic efficacy of another therapeutic agent. The amount of a given composition that will correspond to such amount will vary depending upon a variety of factors, such as the given composition, the pharmaceutical formulation, the route of administration, the condition, the type of disease or disorder, the identity of the subject or host being treated, but nonetheless can be routinely determined by those skilled in the art. As defined herein, the therapeutically effective amount of an agent can be readily determined by the skilled artisan by conventional methods known in the art.
As used herein, the term "age-related disease" refers to a variety of conditions attributable to age-related conditions. These conditions include, but are not limited to, osteoporosis, bone loss, arthritis, arthrosclerosis, cataracts, macular degeneration, metabolic diseases including diabetes, neurodegenerative diseases including alzheimer's and parkinson's diseases, immunosenescence, and heart disease including atherosclerosis and dyslipidemia. The phrase "age-related diseases" also encompasses neurodegenerative diseases such as alzheimer's disease and related disorders, ALS, huntington's disease, parkinson's disease and cancer.
As used herein, the term "autoimmune disease" is intended to mean a disease or disorder resulting from an immune response against a self tissue, organ, or a manifestation thereof or resulting condition. An autoimmune disease may represent a condition caused or exacerbated by the production of autoantibodies reactive with an autoimmune antigen or epitope thereof. The autoimmune disease may be tissue-or organ-specific, or it may be a systemic autoimmune disease. Systemic autoimmune diseases include Connective Tissue Diseases (CTD) such as Systemic Lupus Erythematosus (SLE), systemic sclerosis of mixed connective tissue diseases, Polymyositis (PM), Dermatomyositis (DM) and Sjogren's Syndrome (SS). Other exemplary autoimmune diseases also include rheumatoid arthritis and anti-neutrophil cytoplasmic antibody (ANCA) polyangiitis.
As used herein, the term "genetic disease" refers to a disease caused by an abnormality, such as a mutation, in the nuclear genome. Exemplary genetic diseases include, but are not limited to, early aging syndrome, vorner syndrome, and huntington's disease.
As used herein, the term "cancer" includes, but is not limited to, solid cancers and cancers that are hematological spreading. The terms "cancer" and "cancerous" refer to or describe the physiological condition in mammals that is generally characterized by unrestricted cell growth.
As used herein, the terms "mitochondrial disease or disorder" and "mitochondrial disorder" are interchangeable and refer to a group of conditions caused by genetic or acquired mitochondrial damage that result in energy shortages in those areas of the body. Exemplary organs affected by mitochondrial diseases or disorders include those that consume large amounts of energy, such as the liver, muscles, brain, eyes, ears, and heart. The results are often liver failure, muscle weakness, fatigue, and problems with the heart, eyes, and various other systems.
As used herein, the term "mitochondrial DNA abnormality" refers to a mutation in a mitochondrial gene, the product of which is localized to the mitochondria but not observed in cells of healthy subjects. Exemplary diseases associated with mitochondrial DNA abnormalities include, for example, Chronic Progressive External Ophthalmoplegia (CPEO), pearson Syndrome, cohn-seya Syndrome (Kearns-Sayre Syndrome) (KSS), diabetes with deafness (DAD), Leber's Hereditary Optic Neuropathy (LHON), LHON-plus, neuropathy, ataxia, and retinitis pigmentosa Syndrome (NARP), Maternally Inherited Leigh Syndrome (MILS), also known as Leigh Syndrome caused by mutant mtDNA, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like attacks (MELAS), myoclonic epilepsy with ragged-red fiber disease (MERRF), familial bilateral striatal necrosis/striatal steatosis (FBSN), Luft disease, aminoglycoside-induced deafness (AID), and various deletions of mitochondrial DNA Syndrome.
In the context of a mitochondrial disease or disorder, as used herein, the term "nuclear DNA abnormality" denotes a mutation or change in the coding sequence of a nuclear gene whose product is localized to the mitochondria. Exemplary mitochondrial diseases or disorders associated with nuclear mutations include mitochondrial DNA deletion syndrome-4A, mitochondrial recessive ataxia syndrome (MIRAS), mitochondrial neuro-gastrointestinal encephalomyopathy (MNGIE), mitochondrial DNA deletion syndrome (MTDPS), DNA polymerase gamma (POLG) -related disorders, sensory ataxia neuropathy with dysarthria and ophthalmoplegia (SANDO), leukoencephalopathy with involvement of the brain stem and spinal cord and elevated lactate (LBSL), coenzyme Q10 deficiency, Leigh syndrome (caused by nuclear mutations), mitochondrial complex abnormalities, fumarate deficiency, alpha-ketoglutaradehyde dehydrogenase complex (KGDHC) deficiency, succinyl-coa ligase deficiency, pyruvate dehydrogenase complex deficiency (PDHC), Pyruvate Carboxylase Deficiency (PCD), carnitine palmitoyltransferase i cpt i deficiency (cpt i) deficiency, j, Carnitine palmitoyltransferase ii (cpt ii) deficiency, carnitine-acyl-carnitine (CACT) deficiency, autosomal dominant-/autosomal recessive-progressive extra-ocular paralysis (ad-/ar-PEO), infantile spinocerebellar atrophy (IOSCA), Mitochondrial Myopathy (MM), Spinal Muscular Atrophy (SMA), growth arrest, aminouria, cholestasis, iron overload, early death (GRACILE), and Charcot-Marei-Tooth 2A (CMT 2A).
As used herein, the term "dysfunctional mitochondria" means mitochondria as opposed to functional mitochondria. Exemplary dysfunctional mitochondria include mitochondria that are unable to synthesize ATP or that synthesize an insufficient amount of ATP via oxidative phosphorylation. As used herein, the term "functional mitochondria" refers to mitochondria that consume oxygen and produce ATP.
As used herein, the term "mutation" refers to any change in the structure of a gene that results in the production of a variant (also referred to as "mutant") form. Genetic mutations may be caused by the alternation of single bases in DNA or by deletion, insertion or duplication of larger genetic regions or chromosomes. In some embodiments, the mutation may affect the function or the protein produced. For example, single nucleotide mutations (i.e., point mutations) in DNA in the coding region of a protein can result in the generation of codons that encode different amino acids (i.e., missense mutations). It is understood that such different amino acids may alter the protein structure and, in some cases, the function of organelles, such as mitochondria, as described herein.
As used herein, the terms "heterogeneity" and "heterogeneous" refer to the presence of more than one type of mitochondrial DNA genome in an individual or sample. Different degrees of heterogeneity are associated with different degrees of physiological conditions described herein. Heterogeneity can be identified by means known in the art, and it is expected that the severity of the physiological condition associated with a particular nucleotide allele will vary with the percentage of such associated allele within an individual.
When used in the context of mitochondrial DNA, the term "wild-type", as used herein, refers to the genotype of a typical species form as it exists in nature. An exemplary reference genome for a wild-type human mtDNA genome includes Cambridge Reference Sequence (CRS).
As used herein, the term "old" or "older" is intended to mean that the source of mtDNA is from a subject that is older than the recipient cell or, relative to the recipient cell, from a cell in a cell population that has a greater number of population doublings (i.e., population doubling level, PDL) since they have been cultured ex vivo.
As used herein, the term "young" or "younger" is intended to mean that the source of mtDNA is from a subject that is less aged than the recipient cell or from a cell in a population of cells that has fewer population doublings (i.e., population doubling levels, PDL) since they have been cultured ex vivo relative to the recipient cell.
When used to refer to mitochondria, the term "isolated" as used herein refers to mitochondria that have been physically separated from or removed from other cellular components of their natural biological environment.
As used herein, the terms "intact" and "intact mitochondria" refer to mitochondria that comprise an outer and inner membrane, an intermembrane cavity, a cristae (formed from the inner membrane), and a stroma. An exemplary intact mitochondrion contains mtDNA. In a preferred embodiment, the intact mitochondria are functional mitochondria. However, it is to be understood that intact dysfunctional mitochondria can also be used in the present invention.
As used herein, the term "autologous" is intended to mean a biological component obtained from the same subject.
As used herein, the term "allogeneic" is intended to mean a biological component that is derived from the same species, but is genotypically different from the subject receiving the biological component.
As used herein, the term "animal cell" is intended to mean any cell from a eukaryote. It is understood that animal cells may include mammalian and non-mammalian species, such as amphibians, fish, insects (e.g., Drosophila), and worms (e.g., Caenorhabditis elegans).
As used herein, the term "fusion protein" refers to amino acid sequences that are linked to one another primarily, but not necessarily, by peptide bonds, where one portion of the sequence is derived from (i.e., has sequence similarity to) one source (natural or synthetic) and another portion of the sequence is derived from one or more other sources. Exemplary fusion proteins can be prepared by constructing an expression vector that encodes the entire fusion protein (encoding two parts, e.g., a mitochondrial-targeting sequence and an endonuclease) such that substantially all of the linkages are peptide bonds. It will also be appreciated that fusion may be performed by chemical conjugation, such as by using any known method for conjugating peptides.
As used herein, the terms "mitochondrial-targeting sequence (MTS)" and "mitochondrial-targeting sequence (MTS)" are interchangeable and refer to any amino acid sequence that is capable of causing the transport of an enzyme, peptide, sequence or compound attached thereto into the mitochondria. In certain embodiments, the MTS is a human MTS. In another embodiment, the MTS is from another species. Non-limiting examples of such sequences are cytochrome c oxidase subunit X (COX10) MTS (MAASPHTLSSRLLTGCVGGSVWYLERRT, SEQ ID NO: 36) and cytochrome c oxidase subunit VIII (COX8) MTS (MSVLTPLLLRSLTGSARRLMVPRA, SEQ ID NO: 37). Other non-limiting examples of MTS sequences are the native MTS of each individual mitochondrial protein encoded by nuclear DNA, translated (produced) in the cytoplasm and transported into the mitochondria, as well as citrate synthase (cs), lipoamide dehydrogenase (LAD) and C6ORF66 (ORF). Among them, the various MTSs may be interchangeable for each mitochondrial enzyme. Each possibility represents a separate embodiment of the fusion protein for use in the present invention.
As used herein, the term "small molecule" refers to a compound that affects a biological process and has a molecular weight of about 900 daltons or less. Exemplary small molecules have a molecular weight between about 300 and about 700 daltons.
When used in connection with numerical values, the terms "about" or "approximately" as used herein mean any value within 1, 5, 10, 15, or 20% of the referenced value.
As used herein, the term "somatic cell" refers to any differentiated cell that forms an organism, except stem cells, progenitor cells, and germ cells (i.e., oogonial and spermatogonial cells) and cells derived therefrom (e.g., oocytes, sperm). For example, viscera, skin, bone, blood, and connective tissue are all composed of somatic cells. Somatic cells are obtained from animal, preferably human subjects, and cultured according to standard cell culture procedures available to those skilled in the art.
As used herein, the term "endocytic pathway" refers to a cellular process in which cells take up molecules from their surroundings. The endocytic pathway can be "endostatin-dependent" requiring recruitment of endostatin to help the plasma membrane bend into vesicles that absorb the molecule, or "endostatin-independent" requiring no recruitment of endostatin. Exemplary types of endosytosis that are independent of endosomes include, for example, megapinocytosis. As used herein, the term "endocytic activator" refers to an agent that, for example, induces or activates an endocytic pathway or process, thereby increasing the endocytic pathway. Exemplary "endocytic activators" enhance mitochondrial uptake from the extracellular environment.
As used herein, the term "macroendocytosis" refers to an endosomal form independent of endosomes that mediates non-selective uptake of solute molecules, nutrients, and antigens.
As used herein, the term "compound" refers to a compound capable of eliciting a desired biological function. The term includes, but is not limited to, DNA, RNA, proteins, polypeptides and other compounds, including growth factors, cytokines, hormones or small molecules.
As used herein, the terms "peptide", "polypeptide" and "protein" are used interchangeably and in their broadest sense refer to an amino acid sequence that is constrained (i.e., has some structural elements such as, for example, the presence of an amino acid that causes a β -turn or β -sheet, or, for example, cyclization by the presence of a disulfide-bonded Cys residue) or unconstrained (e.g., linear or unstructured). The amino acids that make up the polypeptide may be of natural origin or may be synthetic. The polypeptide may be purified from a biological sample. Polypeptides, proteins or peptides also encompass modified polypeptides, proteins or peptides, e.g., glycopolypeptides, glycoproteins or glycopeptides; or a lipopeptide, lipoprotein, or lipopeptide.
As used herein, the terms "modulate" and "modulator" are intended to mean a change in the nature or composition of a basal homeostasis state. Exemplary modulation includes altering cellular metabolism by disrupting homeostasis, thereby causing a significant decrease in cellular metabolism. The term "modulator" includes inhibitors and activators. An inhibitor is an agent that, for example, inhibits the expression or modification of a desired protein, pathway or process, or binds to, partially or completely blocks stimulation, reduces, prevents, delays activation, inactivates, desensitizes or downregulates the activity of the target protein, pathway or process. In certain embodiments, the inhibitor is an antagonist of the target protein, pathway or process. An activator is an agent that, for example, induces or activates the expression or modification of the target protein, pathway or process, or binds to, stimulates, increases, opens, activates, facilitates, enhances activation of inhibitor activity, sensitizes or upregulates the activity of the target protein (or encoding polynucleotide), pathway or process. In certain embodiments, the activator is an agonist of the target protein, pathway or process. Modulators include naturally occurring and synthetic ligands, antagonists, and agonists (e.g., small chemical molecules, antibodies, etc., that act as agonists or antagonists). It will also be understood that the modulator may be biological (e.g., an antibody) or chemical.
As used herein, the term "prior" is intended to mean a period of time prior to the beginning of an event such that it is long enough to achieve and maintain a desired result (e.g., antibiotic selection) or effect (e.g., biological effect), while the desired result or effect does not completely dissipate prior to the initiation of the intended event. For example, in an exemplary context, it is understood that modulating cellular metabolism prior to the transfer of exogenous mitochondria and/or exogenous mtDNA will comprise a sufficient period of time to, for example, exhibit a desired biological effect (e.g., an increase in phosphorylation of S6 kinase) while not returning to an homeostasis state prior to the transfer of exogenous mitochondria and/or exogenous mtDNA occurring.
As used herein, the term "nutritional stress" refers to nutrient deficiency or nutrient starvation conditions sufficient to produce perturbations in cellular homeostasis, such as causing autophagy, AMPK signaling, and/or mTOR signaling pathways. Exemplary nutritional stress conditions include serum starvation, essential amino acid removal, and/or metabolic pathway disruption.
The terms "nucleic acid" and "polynucleotide" are used interchangeably herein to describe a polymer of any length composed of nucleotides, e.g., deoxyribonucleotides or ribonucleotides, or synthetically produced compounds that can hybridize to a naturally occurring nucleic acid in a sequence-specific manner similar to two naturally occurring nucleic acids, e.g., can participate in watson-crick base-pairing interactions. As used herein, the term "base" is synonymous with "nucleotide" in the context of a polynucleotide sequence, i.e., a monomeric subunit of a polynucleotide. When used to refer to nucleotides, the abbreviation "a" is intended to refer to adenine (a). When used to refer to nucleotides, the abbreviation "G" is intended to refer to guanine (G). When used to refer to nucleotides, the abbreviation "C" is intended to refer to cytosine (C). When used to refer to nucleotides, the abbreviation "T" is intended to refer to thymine (T).
The term "pharmaceutically acceptable" when used in reference to a carrier is intended to mean a carrier, diluent or excipient that must be compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
The practice of the embodiments provided herein will employ, unless otherwise indicated, conventional molecular biology, microbiology, and immunology methods, which are within the skill of the artisan. These techniques are fully described in the literature. Examples of textbooks that are particularly suitable for reference include the following: sambrook et al, Molecular Cloning A Laboratory Manual, 3 rd edition, Cold Spring Harbor Laboratory, New York (2001); ausubel et al, Current Protocols in Molecular Biology, John Wiley and Sons, Baltimore, MD (1999); glover Master code, DNA Cloning, volumes I and II (1985); gait major edition, Oligonucleotide Synthesis (1984); hames&Higgins, Nucleic Acid Hybridization (1984); hames&Higgins major, Transcription and transformation (1984); freshney eds, Animal Cell Culture: Immobilized Cells and Enzymes (IRL Press,1986);Et al, Plant Molecular Biology-A Laboratory Manual (Main ed. by Mel S.Clark; Springer-Verlag, 1997); immunochemical Methods in Cell and Molecular Biology (Academic Press, London); scopes, Protein Purification: Principles and Practice (Springer Verlag, N.Y., 2 nd Ed. 1987); and Weir &Blackwell's eds, Handbook of Experimental Immunology, Vol.I-IV (1986).
5.2Method for producing mitochondrial-substituted cells (MirC)
The present invention is based in part on the following findings: any agent that reduces endogenous mitochondrial function, including agents that reduce endogenous mitochondrial dna (mtdna), can enhance non-invasive transfer of exogenous mitochondria. However, complete elimination of endogenous mtDNA, such as rho (0) cells, prevents this increase. This is because noninvasive transfer of exogenous mitochondria is energy-dependent, and complete elimination of endogenous mtDNA greatly limits the energy available to assist the noninvasive transfer process. Similarly, non-invasive transfer of exogenous mitochondria is also inefficient when mitochondrial function and/or mtDNA is undisturbed, e.g., when mitochondria are co-incubated (i.e., "appended") or added by centrifugation alone.
Accordingly, provided herein are methods of producing a mitochondrial replaced cell (MirC) which can comprise (a) contacting a recipient cell with an agent that reduces endogenous mtDNA copy number or an agent that reduces mitochondrial function; (b) incubating the recipient cell with the agent for a period of time sufficient to partially reduce endogenous mtDNA copy number or partially reduce endogenous mitochondrial function in the recipient cell, respectively; and (c) co-incubating (1) the recipient cell from step (b) in which endogenous mtDNA or endogenous mitochondrial function, respectively, has been partially reduced and (2) exogenous mitochondria from a healthy donor for a time sufficient to non-invasively transfer the exogenous mitochondria to the recipient cell, thereby producing a mitochondria-replaced cell. Also provided herein is a method of producing a mitochondrial-replaced cell comprising performing steps (a) and (b) as described above, and then (c) co-incubating (1) the recipient cell from step (b) in which endogenous mtDNA or endogenous mitochondrial function, respectively, has been partially reduced and (2) exogenous mtDNA from a healthy donor for a period of time sufficient to non-invasively transfer exogenous mtDNA to the recipient cell, thereby producing a mitochondrial-replaced cell. In certain embodiments, the exogenous mtDNA is transferred by an exogenous mitochondrion.
The generation of MirC can be a useful strategy for a variety of applications. For example, transfer of exogenous mitochondria, exogenous mtDNA, or a combination thereof to a recipient cell can be used, for example, to replace dysfunctional endogenous mitochondria and/or endogenous mitochondria composed of mutant mtDNA having functional mitochondria, such as mitochondria composed of wild-type mtDNA. In certain embodiments, the methods provided herein are performed in a recipient cell having endogenous mtDNA encoding a dysfunctional mitochondrion. In a specific embodiment, the endogenous mtDNA is mutant mtDNA. In certain embodiments, the endogenous mtDNA is heterogeneous and consists of both wild-type mtDNA and mutant mtDNA.
As described above, in certain applications, the transfer of exogenous mitochondria, exogenous mtDNA, or a combination thereof can include the transfer of functional mitochondria or wild-type mtDNA to replace, for example, dysfunctional or endogenous mitochondria consisting of mutant mtDNA. Thus, in certain embodiments, the exogenous mtDNA is wild-type mtDNA. In other embodiments, the endogenous mitochondria of the recipient cell have wild-type mtDNA and dysfunctional endogenous mitochondria. For example, exemplary dysfunctional mitochondria of recipient cells with wild-type mtDNA can include mutant nuclear DNA encoding mitochondrial proteins or dysfunctional mitochondria due to secondary effects such as aging or disease.
Thus, a dysfunctional endogenous mitochondrion, an endogenous mitochondrion consisting of mutant mtDNA, or a combination thereof can be replaced using the methods described herein. Mitochondrial dysfunction can occur due to a variety of factors. Non-limiting examples include mitochondrial dysfunction due to a disease (e.g., an age-related disease, a mitochondrial disease or disorder, a neurodegenerative disease, a retinal disease, a genetic disease), diabetes, an auditory disorder, or any combination thereof. Mitochondrial dysfunction can include a reduction in endogenous mitochondrial function of greater than 5%, greater than 10%, greater than 20%, greater than 30%, greater than 40%, greater than 50%, greater than 60%, greater than 70%, greater than 80%, or greater than 90%. Thus, in some embodiments, endogenous mitochondria comprise mitochondria that have a reduced function by about 5%, about 10%, about 15%, about 20%, about 25%, about 30%, about 40%, about 50%, about 60%, about 70%, about 80%, about 90%, or about 100%.
The methods provided herein are applicable to both homogenous and heterogeneous mtDNA. In a specific embodiment, the endogenous mtDNA is a single type of mtDNA (i.e., the endogenous mtDNA is homogeneous). In other specific embodiments, the endogenous mtDNA comprises more than one type of mtDNA (i.e., the endogenous mtDNA is heterogeneous). In some embodiments, the heterogeneous mtDNA comprises both wild-type mtDNA and mutant mtDNA. Generally, the proportion of mutant mtDNA determines the severity of the phenotype and can affect the extent of reduced mitochondrial function. For example, in some embodiments, the heterogeneous mtDNA is 5% mutant mtDNA and 95% wild-type mtDNA, and mitochondrial function is reduced by 5%. In other embodiments, the heterogeneous mtDNA is 55% mutant mtDNA and 45% wild-type mtDNA, and mitochondrial function is reduced by 55%. However, it is understood that the percentage of mutant mtDNA need not be directly proportional to mitochondrial function.
Dysfunctional mitochondria are generally characterized by reduced efficiency of the electron transport chain and reduced synthesis of high energy molecules, such as adenosine-5' -triphosphate (ATP), leakage of harmful Reactive Oxygen Species (ROS), and/or destruction of cellular respiration. One skilled in the art will understand how to assess mitochondrial function. For example, cell-based assays, such as the Seahorse Bioscience XF Extracellular Flux Analyzer (excellar Flux Analyzer), can be used to perform assays of basal oxygen consumption, glycolysis rate, ATP production, and respiration volume in a single experiment to assess mitochondrial dysfunction. Similarly, the orobos 02K respirometer can also be used to establish quantitative functional mitochondrial diagnosis. It is to be understood that the assay examples described above are exemplary and do not include all methods of assessing mitochondrial function.
In some embodiments, the functional mitochondria have an intact outer membrane. In some embodiments, the functional mitochondria are intact mitochondria. In another embodiment, the functional mitochondria consume oxygen at an increased rate over time. In another embodiment, mitochondrial functionality is measured by oxygen consumption. In another embodiment, the oxygen consumption of mitochondria can be measured by any method known in the art, such as, but not limited to, MitoXpress fluorescent probe (Luxcel). In some embodiments, the functional mitochondria are mitochondria that exhibit an increased rate of oxygen consumption in the presence of ADP and a substrate, such as (but not limited to) glutamate, malate, or succinate. Each possibility represents a separate embodiment of the invention. In another embodiment, the functional mitochondria are ATP-producing mitochondria.
Although the methods provided herein can be used to produce MirC from recipient cells having dysfunctional mitochondria, mutant mtDNA, or a combination thereof, it is also understood that production of MirC need not be performed in recipient cells having dysfunctional mitochondria. In some embodiments, the MirC is produced using recipient cells having functional endogenous mitochondria, wild-type mtDNA, or a combination thereof, and the exogenous mitochondria are also functional, containing wild-type mtDNA, or a combination thereof. For example, endogenous wild-type mtDNA can be reduced using the methods provided herein, and exogenous wild-type mtDNA can be transferred into a recipient cell, such as mitochondrial replacement in an "old" recipient cell (e.g., a cell from an aging subject or a cell with a relatively high Population Doubling Level (PDL)) using exogenous mtDNA from a healthy donor cell (e.g., a young cell with a relatively low PDL). Thus, in certain embodiments, the exogenous mtDNA is from a donor cell that is a healthy donor cell, e.g., a younger donor cell than the recipient cell. In certain embodiments, the PDL difference between the donor and recipient cells is about 1.5 fold, about 2 fold, about 2.5 fold, about 3 fold, about 4 fold, about 5 fold, or greater than 5 fold. In other embodiments, the donor and recipient cells are from subjects that are about 1.5-fold, about 2-fold, about 2.5-fold, about 3-fold, about 4-fold, about 5-fold, or greater than 5-fold different in age. However, it is understood that age differences between donor and recipient cells are not required. In some embodiments, the donor and recipient cells are of the same age, and the donor cell is a healthy cell.
In other embodiments, production of MirC is performed in recipient cells having a functional endogenous mitochondrion, such as a wild-type endogenous mtDNA, and the exogenous mtDNA is mutated, encodes a dysfunctional mitochondrion, is dysfunctional or a combination thereof. In other embodiments, the exogenous mitochondrion, exogenous mtDNA, or a combination thereof is from a donor cell that is older than the recipient cell. For example, in some embodiments, a mitochondrial disease or disorder model can be generated by replacing a functional mitochondrion in a recipient cell with exogenous mtDNA from a donor cell that is mutated and/or encodes a dysfunctional mitochondrion. It is understood that the examples described herein are exemplary and do not include all combinations involving mtDNA substitutions.
As provided herein, methods of producing MirC can be practiced using agents that reduce endogenous mtDNA or agents that reduce endogenous mitochondrial function. In some cases, a combination of two agents may be used. Agents capable of reducing mitochondrial function are well known in the art and are within the skill of those in the art. Exemplary agents include mitochondrial respiratory chain inhibitors that block respiration in the presence of ADP or uncouplers, such as complex III inhibitors (e.g., mucothiazole), complex IV inhibitors (e.g., sodium azide, potassium cyanide (KCN)), or complex V inhibitors (e.g., oligomycin); phosphorylation inhibitors that eliminate oxygen consumption bursts after ADP addition, but have no effect on uncoupler-stimulated respiration; uncouplers that eliminate the necessary linkage between the respiratory chain and the phosphorylation system that are observed to have intact mitochondria (e.g., dinitrophenol, CCCP, FCCP); inhibitors of ATP/ADP transport that prevent ATP export or import of raw materials across the inner mitochondrial membrane, such as adenine nucleotide translocase inhibitors (e.g., atractyloside); ionophores that render the inner membrane permeable to compounds that are not normally able to cross the membrane (e.g., valinomycin, nigericin); or a TCA cycle inhibitor (e.g., arsenite, aminooxyacetate) that blocks one or more TCA cycle enzymes or ancillary reactions. It is to be understood that the agents capable of reducing mitochondrial function as described above are non-limiting and that one skilled in the art can readily identify suitable agents capable of reducing mitochondrial function using techniques known in the art.
In particular embodiments, the agent that reduces endogenous mitochondrial function transiently reduces endogenous mitochondrial function. In other embodiments, the agent that reduces endogenous mitochondrial function permanently reduces endogenous mitochondrial function. In preferred embodiments, the agent that reduces endogenous mitochondrial function partially reduces endogenous mitochondrial function.
Various reagents can be used to reduce mtDNA. In certain embodiments, the agent that reduces mtDNA is selected from a nucleic acid encoding a fusion protein comprising a mitochondrial-targeting sequence (MTS) and an endonuclease, endonuclease or small molecule. In certain embodiments, the small molecule is a Nucleoside Reverse Transcriptase Inhibitor (NRTI). The nucleic acid may be a messenger ribonucleic acid (mRNA) or a deoxyribonucleic acid (DNA). In certain embodiments, the mtDNA-reducing agent is a plasmid DNA expression vector cassette encoding an endonuclease. In a preferred embodiment, the agent is a plasmid DNA expression vector cassette encoding an endonuclease with MTS. A variety of expression vector cassettes can be used, and one skilled in the art will appreciate the necessary considerations required to enable successful expression of the endonuclease based on the host cell. For example, a mammalian expression vector, such as a vector having a Cytomegalovirus (CMV) promoter, SV40 promoter or CAG promoter, would be suitable for expression of the endonuclease in mammalian cells, but not non-mammalian cells. Similarly, it will be understood that viral expression vectors may also be used and those skilled in the art will appreciate that these viral expression vectors may require helper plasmids (i.e., envelope and packaging plasmids) to be used in tandem with the transfer plasmid. In other embodiments, the agent is mRNA encoding an endonuclease. In other preferred embodiments, the agent is mRNA encoding an endonuclease having an MTS. In other embodiments, the agent is an endonuclease that is a recombinant protein. In other embodiments, the agent is a small molecule, such as, for example, a small molecule that disrupts mtDNA synthesis. Techniques for generating any expression method are known to those of skill in the art and can be readily implemented without undue experimentation. In a preferred embodiment, the agent is suitable for clinical use.
In a specific embodiment, the endonuclease can be a restriction endonuclease that cleaves the DNA duplex into fragments at specific sites, such as XbaI, that cleaves the following DNA sequences:
the endonuclease may also include, for example, restriction endonucleases other than XbaI, such as EcoRI, BamHI, HindIII or PstI, each of which digests mtDNA at multiple sites. Endonucleases have defined recognition sites which allow their sensitivity to mtDNA to be predicted. Defined recognition sites for restriction endonucleases, such as, for example, XbaI, EcoRI and SmaI, are specific for a given nucleic acid sequence. Thus, in some embodiments, the reduction of endogenous mtDNA can be performed using zinc fingers and transcription activator-like effectors (TALEs) that have been incorporated into DNA nucleases. Both types of DNA-binding proteins can be engineered to be specific for a new DNA sequence of interest. Similarly, Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 protein can also be introduced into cells by adding the corresponding encoding genes. Thus, in some embodiments, the endonuclease can be a programmable nuclease, such as an RNA-guided DNA endonuclease (e.g., Cas9), a Zinc Finger Nuclease (ZFN), or a transcription activator-like effector nuclease (TALEN). It is understood that nucleases as described above are non-limiting and that a person skilled in the art can readily identify suitable endonucleases using techniques known in the art. For example, a cambridge reference sequence or similar consensus sequence can be analyzed, e.g., in silico, to identify suitable endonucleases for identifying mtDNA sequences. In a specific embodiment, the endonuclease cleaves a wild-type mtDNA sequence. In other embodiments, the endonuclease cleaves a mutant mtDNA sequence. It will also be understood that the agent that reduces endogenous mtDNA need not be an endonuclease and that any agent capable of reducing mtDNA may be used, including agents that inhibit mtDNA biosynthesis, such as ethidium bromide. Also contemplated herein are agents such as, for example, urolithin a or small molecule p 62-mediated mitophagy inducers (PMIs) that induce autophagy to promote selective degradation of endogenous mitochondria (i.e., mitophagy agonists). The present invention can also be practiced using Nucleoside Reverse Transcriptase Inhibitors (NRTIs) as agents to reduce mtDNA.
In addition, in some embodiments, the expression vector cassette may include one or more antibiotic resistance genes to enable selection of a population of cells expressing the expression vector cassette. For example, in some embodiments, the expression vector can include a puromycin N-acetyl-transferase gene (pac) from streptomyces, and cells can be selected using puromycin. In the case where the selection is performed using an antibiotic, e.g., puromycin, the selection can be brief (e.g., 24-48 hours) to limit long term exposure to the drug. However, it is to be understood that the examples provided above are merely exemplary and that the expression vector cassette may include other antibiotic resistance genes, such as, for example, the bsr, bls or BSD genes for selection by blasticidin, or the hph gene for selection by hygromycin B. It will generally be understood that the concentration of antibiotic used for selection will be based on the type of antibiotic and the cell type, and is readily available to those skilled in the art without undue experimentation. It will also be understood that selection may be generated by any means known in the art and need not include antibiotic resistance. For example, in some embodiments, cell selection can be performed by, for example, Fluorescence Activated Cell Sorting (FACS) of cell surface markers or by expression of the fluorescent protein encoded by the expression. In other embodiments, selection may be based on cell phenotype. For example, in some embodiments, successful deletion of mutant endogenous mtDNA in a cell with heterogeneity can result in a selectable phenotypic response, such as (for example) cell survival.
Thus, in some embodiments, the cell is selected after introduction of the expression vector cassette containing the mtDNA-degrading endonuclease. In some embodiments, the cells are selected to obtain a uniform population of cells expressing an mtDNA-degrading endonuclease. In a specific embodiment, cells are selected after introduction of an expression vector cassette containing an mtDNA-degrading endonuclease and a uniform stable cell line is generated. In other embodiments, the cells are selected to enrich for a population of cells that express an mtDNA-degrading endonuclease. As mentioned above, this enrichment by selection may include a brief exposure to antibiotics. The enriched cells may stably express the endonuclease or transiently express the endonuclease based on the degree and/or manner of selection pressure. It is understood that the enriched population need not be homogeneous, and that this enriched cell population expressing an mtDNA-degrading endonuclease contains a higher proportion of cells having the endonuclease relative to the unselected cell population, but may also contain some cells that do not express the endonuclease.
In other embodiments, the cell is not selected after introduction of the expression vector comprising an mtDNA-degrading endonuclease. In a specific embodiment, the cell is not selected and the endonuclease is transiently expressed after introduction of the expression vector containing the mtDNA-degrading endonuclease.
Various methods for introducing plasmid DNA expression vector cassettes, mRNA and/or recombinant proteins are known in the art. In some embodiments, the plasmid DNA expression vector cassette is introduced by electroporation. In a specific embodiment, the electroporation method is flow electroporation, such as MaxCyte flow electroporation. In other specific embodiments, electroporation involves nuclear transfection techniques, such as the Nucleofector of LonzaTMProvided is a technique. In other embodiments, the plasmid DNA expression vector cassette is introduced by cationic lipofection. In other embodiments, the plasmid DNA expression vector cassette is introduced by viral transduction. It is to be understood that the methods for introducing an expression vector cassette as described above are non-limiting and are intended to be exemplary methods only, and that any method known in the art may be used for introducing a DNA expression vector cassette.
When the agent for reducing endogenous mitochondria comprises an endonuclease, expression of the endonuclease can also include introduction of mRNA encoding the endonuclease or introduction of the endonuclease as a recombinant protein. In certain embodiments, the MaxCyte electrotransformer may be used for mRNA transfection, particularly in a clinical setting, which has passed the standards of good manufacturing practice and good clinical practice. Transfection may be performed using a MaxCyte electrotransformation machine, according to the manufacturer's protocol. It will also be appreciated that the methods described above are merely exemplary and that any manner of introducing mRNA and/or recombinant protein may be used.
Specific targeting of mitochondria by an endonuclease can be performed by introducing a Mitochondrial Targeting Sequence (MTS) adjacent to the endonuclease coding sequence, which will result in the production of a fusion protein targeted to mitochondria. Strong MTS has been identified and when fused at their N-terminus it was shown to be able to target proteins to specific compartments and is referred to as a mitochondrial targeting sequence. MTSs suitable for the methods described herein are well known to those skilled in the art (see, e.g., U.S. patent No.8,039,587B2, which is incorporated herein by reference in its entirety). For example, MTS directed against the mitochondrial matrix, such as MTS targeting peptides from cytochrome c oxidase subunit IV (COX 4), subunit VIII (COX 8), or subunit X (COX 10) may be used. In principle, any target or artificial sequence derived from any nuclear-encoded mitochondrial matrix or endomembrane enzyme (hydrophobic moment greater than 5.5, at least two basic residues, amphipathic α -helical conformation; see, e.g., Bedwell et al, Mol Cell biol.9(3) (1989),1014-1025) that is capable of making the fusion protein a mitochondrially transported protein is useful for the purposes of the present invention.
In certain embodiments, the MTS is a human MTS. In another embodiment, the MTS is from another species. Non-limiting examples of these sequences are cytochrome c oxidase subunit X (COX 10) MTS (MAASPHTLSSRLLTGCVGGSVWYLERRT, SEQ ID NO: 36) and cytochrome c oxidase subunit VIII (COX 8) MTS (MSVLTPLLLRSLTGSARRLMVPRA, SEQ ID NO: 37). Other non-limiting examples of MTS sequences are the native MTS of each individual mitochondrial protein encoded by nuclear DNA, translated (produced) in the cytoplasm and transported into the mitochondria, as well as citrate synthase (cs), lipoamide dehydrogenase (LAD) and C6ORF66 (ORF). Among them, the various MTSs may be interchangeable for each mitochondrial enzyme. Thus, in some embodiments, the MTS targets mitochondrial matrix proteins. In a specific embodiment, the mitochondrial matrix protein is subunit VIII of human cytochrome C oxidase. Each possibility represents a separate embodiment of the fusion protein for use in the present invention.
Once a recipient cell is contacted with an agent that reduces endogenous mtDNA copy number or an agent that reduces endogenous mitochondrial function, the recipient cell is incubated with the agent for a period of time sufficient to partially reduce endogenous mtDNA copy number or partially reduce endogenous mitochondrial function in the recipient cell, respectively. It is within the skill of the art to recognize "a sufficient period of time" to allow the agent to partially reduce endogenous mtDNA copy number or partially reduce endogenous mitochondrial function. The sufficient or appropriate time period will vary depending on a number of factors including, but not limited to, the particular type of cell, the amount of starting material (e.g., the number of recipient cells and/or the amount of mtDNA to be reduced), the amount and type of agent, the plasmid promoter regulator and/or the culture conditions. In various embodiments, the period of time sufficient to allow a partial reduction in the copy number of endogenous mtDNA in the recipient cell is about 1 day, about 2 days, about 3 days, about 4 days, about 5 days, about 6 days, about 7 days, about 8 days, about 9 days, about 10 days, about 1-2 weeks, about 2-3 weeks, or about 3-4 weeks. In preferred embodiments, the sufficient period of time will be long enough such that the recipient cell produced has a majority of the reduction in copy number of the endogenous mtDNA or a majority of the reduction in endogenous mitochondrial function and is also substantially free of an agent that reduces endogenous mtDNA or an agent that reduces endogenous mitochondrial function prior to incubating the recipient cell with exogenous mtDNA and/or exogenous mitochondria.
Important and novel aspects of the present invention are the following findings: mitochondrial transfer efficiency was severely reduced in cells with complete elimination of inner mitochondria (i.e., (p) 0 cells),but when the endogenous mtDNA copy number is reduced but not completely eliminated (i.e., (ρ)-Cells), can be greatly improved. Furthermore, the present invention also demonstrates that simple addition or centrifugation protocols are ineffective, which do not partially reduce endogenous mtDNA copy number. Thus, in a preferred embodiment, the reduction in copy number of endogenous mtDNA in the recipient cell is less than 100% elimination of endogenous mtDNA. In some embodiments, the endogenous mtDNA copy number in the recipient cell is reduced by about 5% to about 99%. In a specific embodiment, the agent that reduces endogenous mtDNA copy number by about 30% to about 70%. In other embodiments, the agent that reduces the copy number of endogenous mtDNA by about 50% or more, about 60% or more, about 70% or more, about 80% or more, or about 90% or more, or about 95% or more. In other embodiments, the agent that reduces endogenous mtDNA copy number by about 60% to about 90%. It will also be appreciated that in some embodiments, the agent that reduces the copy number of endogenous mtDNA reduces mitochondrial material.
In certain embodiments, the exogenous mtDNA is contained in an isolated exogenous mitochondrion from a donor cell. Isolation of mitochondria can be achieved by any of a number of well-known techniques, including, but not limited to, those described herein and in the cited references. In certain embodiments, exogenous mitochondria for use in mitochondrial transfer are isolated using commercially available kits, such as, for example, the Qproteum mitochondrial isolation kit (Qiagen, USA), the MITOI 2 mitochondrial isolation kit (Sigma, USA), or the mitochondrial isolation kit for cultured cells (Thermo Scientific). In other embodiments, the exogenous mitochondria used for use in mitochondrial transfer are isolated manually. For example, an exemplary manual isolation of mitochondria involves washing about 10 from growing in culture by granulating donor cells91-2mL of cell pellet of individual cells, swelling the cells in hypotonic buffer, disrupting the cells using a closely-fitted pestle with a Dounce or Potter-Elvehjem homogenizer and separating the mitochondria by differential centrifugation, separating from the donor cellsOff-mitochondria. Manual separation may also include, for example, sucrose density gradient ultracentrifugation or free flow electrophoresis. Without wishing to be bound by any particular method, it is understood that the kits and manual methods described herein are exemplary and that any mitochondrial isolation method can be used, and will be within the skill of one of skill in the art.
In some embodiments, the isolated donor mitochondria are substantially free of other organelles. In other embodiments, the isolated mitochondria can contain impurities and be enriched for mitochondria. For example, in some embodiments, the isolated mitochondria are about 90% pure, about 80% pure, about 70% pure, about 60% pure, about 50% pure, or any integer therebetween. In general, it will be appreciated that once mitochondria are transferred, any impurities contained with the isolated donor mitochondria will not affect the viability or function of the recipient cell. In particular embodiments, the transfer of exogenous mitochondrial, exogenous mtDNA, or a combination thereof does not include transfer of non-mitochondrial organelles.
The quantity and quality of the isolated mitochondria can be readily determined by a number of well-known techniques, including, but not limited to, those described herein and in the cited references. For example, in some embodiments, the amount of isolated mitochondria is determined by an assessment of total protein content. Various methods are available for measurement of total protein content, such as the biuret and Lowry procedures (see, e.g., Hartwig et al, Proteomics,2009 Jun; 9(11): 3209-14). In other embodiments, the amount of mitochondria isolated is determined by mtDNA copy number.
In some embodiments, the isolated mitochondria are functional mitochondria. In other embodiments, the isolated mitochondria are dysfunctional mitochondria. In some embodiments, mitochondrial function in the donor cell can be assessed prior to isolation. In other embodiments, mitochondrial function can be determined from isolated mitochondria.
Maintenance of mitochondrial membrane integrity is another important factor during mitochondrial isolation. In some embodiments, the mtDNA used in the methods provided herein is from intact mitochondria. In specific embodiments, about 10%, about 20%, about 30%, about 40%, about 50%, about 60%, about 70%, about 80%, about 90%, or greater than 90% of the isolated mitochondria are intact. Integrity of the mitochondrial membrane can be achieved by any of a number of well-known techniques, including, but not limited to, those described herein and in the cited references. For example, TMRM, Rhod123, JC-1 and DiOC6 are typical probes used to measure mitochondrial membrane potential (see, e.g., Perry et al, Biotechniques,2011 Feb; 50(2): 98-115). JC-1 is a widely used dye for measuring the inner membrane potential of isolated mitochondria and is based on the electrochemical proton gradient of the inner mitochondrial membrane.
In certain embodiments of the methods provided herein, a recipient with a reduction in endogenous mtDNA moieties is co-incubated with exogenous mitochondria from a healthy donor for a sufficient period of time to non-invasively transfer the exogenous mitochondria to the recipient cell, thereby producing a mitochondria-replaced cell. In other embodiments, a recipient with a reduction in endogenous mtDNA portion is co-incubated with exogenous mtDNA from a healthy donor for a period of time sufficient to non-invasively transfer exogenous mitochondria to the recipient cell, thereby producing a mitochondrial-replaced cell. It is within the skill of the art to recognize "a sufficient period of time" to non-invasively transfer exogenous mitochondrial and/or exogenous mtDNA to recipient cells. The sufficient or appropriate time period will vary depending on a number of factors including, but not limited to, the particular type of cell, the amount of starting material (e.g., recipient cell number and/or amount of endogenous mtDNA to be replaced), the amount of donor material (e.g., amount, quality and/or purity of exogenous mtDNA), and/or the culture conditions. In various embodiments, the period of time sufficient for non-invasive transfer of the exogenous mitochondrial and/or exogenous mtDNA to the recipient cell is about 1 day, about 2 days, about 3 days, about 4 days, about 5 days, about 6 days, about 7 days, about 8 days, about 9 days, about 10 days, about 1-2 weeks, about 2-3 weeks, or about 3-4 weeks. In certain embodiments, at the end of the co-incubation period, the recipient cell will have most of the exogenous mtDNA and be substantially free of any exogenous mitochondrial organelles.
Another feature of the present invention resides in the following findings: total mtDNA copy number in MirC is not substantially increased relative to primary recipient cells. In contrast, other less efficient methods have been attempted to append mitochondria without conditioning recipient cells prior to the co-incubation step, or to transfer exogenous mitochondria using centrifugation without conditioning recipient cells prior to centrifugation. Thus, cell populations obtained using null methods tend to have a substantial increase in total mtDNA copy number. Thus, in certain embodiments, the mitochondrially replaced cell has a total mtDNA copy number relative to the total mtDNA copy number of the recipient cell prior to contact with the agent that reduces endogenous mtDNA copy number of no greater than about 1.1 fold, about 1.2 fold, about 1.3 fold, about 1.4 fold, about 1.5 fold or more.
The use of non-invasive transfer is another unique aspect of the present invention. The above methods use traumatic instruments to inject exogenous mitochondria, physically force mitochondria into cells by centrifugation or use similar harsh conditions that are harmful to recipient cells. In the clinical setting, especially when the recipient cell number may be limited, such as with hematopoietic stem cells or T cells, harsh cell manipulation is undesirable. Thus, the use of non-invasive transfer is an advantageous feature of the invention, which facilitates its use in a clinical setting.
As provided herein, the exogenous mitochondria, exogenous mtDNA, or a combination thereof can be autologous or allogeneic to the recipient cell. In some embodiments, the exogenous mtDNA is allogeneic with respect to the recipient cell. For example, the exogenous mtDNA can be derived from the same species as the recipient cell and have a different genotype than the recipient cell. In other embodiments, the exogenous mitochondrial, exogenous mtDNA, or a combination thereof is autologous. For example, exemplary autologous exogenous mtDNA can include mtDNA from a healthy donor cell, e.g., a "young" donor cell, such as a "young" donor cell from umbilical cord blood, and the recipient cell can be from the same subject and can be an "old" recipient cell, where the terms "young" and "old" represent the total number of doublings of cells in the population or the age of the subject from which the cells are derived. Another exemplary autologous exogenous mtDNA may include, for example, donor mtDNA isolated from the same subject as the recipient cell and modified prior to its replacement with the recipient cell. In certain embodiments, only mtDNA and/or mitochondria are allogeneic and the recipient cell is autologous to the subject in need of the exogenous mtDNA and/or exogenous mitochondria.
In certain embodiments, replacement of mtDNA in a recipient cell can be assessed by sequencing a hypervariable region (HVR) DNA sequence of the mtDNA, e.g., HV1 and/or HV2 of the D-loop and comparing it to sequences of both the donor mitochondria and the recipient cell. In particular embodiments, sequence differences between the recipient cell and the donor mitochondria can be identified by single nucleotide polymorphism assays. For example, amplified sequences of mtDNA from recipient cells and donor mitochondria can be cloned into plasmids for use as a quantitative standard.
In some embodiments, the cells (i.e., donor cells and recipient cells) are animal cells or plant cells. In a specific embodiment, the cell is a mammalian cell. In some embodiments, the cells are isolated from a mammalian subject selected from the group consisting of: human, horse, dog, cat, mouse, rat, cow, and sheep. In some embodiments, the cell is a human cell. In some embodiments, the cell is a cell in culture. The cells may be obtained directly from a mammal (preferably a human) or from a commercial source, or from tissue, or in the form of, for example, cultured cells, prepared in situ or purchased from a commercial cell source, and the like. In certain embodiments, the cell is a primary cell (i.e., a cell obtained directly from a living tissue, e.g., biopsy material). The cells may be from any organ, including but not limited to the blood or lymphatic system, from muscle, any organ, gland, skin or brain. In certain embodiments, the cell is a somatic cell. In some embodiments, the cell is selected from the group consisting of an epithelial cell, a neural cell, an epidermal cell, a keratinocyte, a hematopoietic cell (e.g., a bone marrow cell), a melanocyte, a chondrocyte, a hepatocyte, a B cell, a T cell, a erythrocyte, a macrophage, a monocyte, a fibroblast, a muscle cell, a vascular smooth muscle cell, a hepatocyte, a spleen cell, and a pancreatic beta cell.
As provided herein, in particular embodiments, the donor cell is a commercially available cell culture under modern good manufacturing criteria (cGMP). For example, the donor cells can be obtained from a cell bank, such as Waisman Biomanufacturing, or similar commercial sources, such as commercial sources that produce cGMP-compliant cells. In some embodiments, the donor cell is a cGMP-producing bone marrow-derived mesenchymal stromal cell (BM-MSC). In other embodiments, the cell is a cGMP grade human hepatocyte. As such, it will also be understood that the donor cell may be a frozen cell that is thawed prior to isolation of the mitochondria. However, the mitochondria need not be isolated after the cells are frozen and can be isolated from fresh cells and used immediately, or in certain embodiments, can be isolated and then frozen prior to transfer to recipient cells.
In some embodiments, the cell is a cancer cell. Typically, the cancer cell is isolated from a cancer selected from the group consisting of: breast cancer, prostate cancer, lymphoma, skin cancer, pancreatic cancer, colon cancer, melanoma, malignant melanoma, ovarian cancer, brain cancer, primary brain cancer, head and neck cancer, glioma, glioblastoma, liver cancer, bladder cancer, non-small cell lung cancer, head or neck cancer, breast cancer, ovarian cancer, lung cancer, small cell lung cancer, Wilms' tumor, cervical cancer, testicular cancer, bladder cancer, pancreatic cancer, gastric cancer, colon cancer, prostate cancer, genitourinary cancer, thyroid cancer, esophageal cancer, myeloma, multiple myeloma, adrenal cancer, renal cell carcinoma, endometrial cancer, adrenal cortex cancer, malignant islet adenoma, malignant carcinoid carcinoma, choriocarcinoma, Alibecell disease, malignant hypercalcemia, cervical hyperplasia, leukemia, acute lymphocytic leukemia, chronic myelocytic leukemia, Acute myelogenous leukemia, chronic myelogenous leukemia, hairy cell leukemia, neuroblastoma, rhabdomyosarcoma, Kaposi's sarcoma, splenomegaly polycythemia, essential thrombocythemia, Hodgkin's disease, non-Hodgkin's lymphoma, Gentism sarcoma, osteogenic sarcoma, essential macroglobulinemia, and retinoblastoma.
In some embodiments, the cell is a stem cell. As used herein, the term "stem cell" refers to an undifferentiated cell that can induce proliferation. Stem cells are capable of self-maintenance or self-renewal, meaning that by each cell division, one daughter cell will also be a stem cell. The stem cells may be obtained from embryonic, postpartum, juvenile or adult tissues. The stem cells may be pluripotent or multipotent. As used herein, the term "progenitor cell" refers to an undifferentiated cell that is derived from a stem cell and is not itself a stem cell. Some progenitor cells can produce progeny that are capable of differentiating into more than one cell type. Stem cells include pluripotent stem cells, which can form cells of any body tissue lineage: mesoderm, endoderm and ectoderm. Thus, for example, the stem cells may be selected from human Embryonic Stem (ES) cells; human Inner Cell Mass (ICM)/ectodermal cells; human primitive ectodermal cells, human primitive endodermal cells; human primitive mesoderm cells; and human primitive Embryo (EG) cells. Stem cells also include pluripotent stem cells, which can form multiple cell lineages that make up the entire tissue, such as (but not limited to) hematopoietic stem cells or neural precursor cells. Stem cells also include totipotent stem cells, which can form a whole organism. In some embodiments, the stem cell is a mesenchymal stem cell. For adult cells that are not terminally differentiated, the terms "mesenchymal stem cells" or "MSCs" are used interchangeably, based on a variety of influences from biologically active factors, such as cytokines, that can divide to obtain stem cells or irreversibly differentiate to produce mesenchymal lineage cells, e.g., adipose, bone, cartilage, elastic and fibrous connective tissue, myoblasts), and tissues other than those originating from embryonic mesoderm (e.g., neural cells). In some embodiments, the stem cell is a partially differentiated or differentiated cell. In some embodiments, the stem cell is an Induced Pluripotent Stem Cell (iPSC), which has been reprogrammed or dedifferentiated. In a specific embodiment, the recipient cell is an iPSC. In other embodiments, the recipient cell is a Hematopoietic Stem Cell (HSC) or MSC. The stem cells may be derived from embryonic, fetal or adult tissue.
In other embodiments, the cell is an immune cell. In a specific embodiment, the recipient cell is an immune cell. In some embodiments, the immune cell is selected from the group consisting of a T cell, a phagocyte, a microglia, and a macrophage. In a specific embodiment, the T cell is a CD4+ T cell. In other embodiments, the T cell is a CD8+ T cell. In other embodiments, the T cell is a Chimeric Antigen Receptor (CAR) T cell. In particular embodiments, the recipient cell is a depleted or near-depleted T cell in or near a T cell dysfunctional state.
5.3Method for improving mitochondrial transfer
Also provided herein are methods for mtDNA and/or mitochondrial transfer comprising the use of a second active agent in combination with any of the methods described in section 5.2. Mitochondrial transfer has been reported to involve an endocytic pathway, which is an ATP-dependent process. For example, under certain Cell culture conditions, mitochondria have been observed to be engulfed by macroendocytosis (see, e.g., Kitani et al, J Cell Mol Med.,2014,18(8):1694 1703). Thus, the present invention also relates to the following novel discoveries: the use of a second active agent can facilitate uptake of the exogenous mitochondrial and/or exogenous mtDNA prior to co-incubation of the recipient cell with the exogenous mitochondrial and/or exogenous mtDNA.
Various types of agents can be used to promote uptake of exogenous mitochondrial and/or exogenous mtDNA. In some embodiments, the second active agent is selected from a macromolecule, a small molecule, or a cell therapy, and the second active agent is optionally selected from rapamycin, NR (nicotinamide ribose), bezafibrate, idebenone, mercaptoethylamine bitartrate (RP103), elamipramide (MTP131), omaviralone (omavelolone) (RTA408), KH176, vantiquone (Vatiquinone) (Epi743), lipoic acid, a0001 (alpha-tocopherols), mitochondrial CoQ10(MitoQ), SkQ1(Visomitin), resveratrol, curcumin, a ketonic therapy, hypoxia, and an endocytic activator. In a specific embodiment, the endocytic activator is a modulator of cellular metabolism. Cellular metabolism can be regulated using a variety of methods known to those skilled in the art. In certain embodiments, the modulation of cellular metabolism comprises nutrient starvation, a chemical inhibitor, or a small molecule.
As mentioned above, transfer of intact mitochondria has been reported to occur via the endocytic pathway. For example, the exogenous mitochondrial and/or exogenous mtDNA can be transferred by uptake by intact mitochondria via the endocytic pathway. Endocytic pathways can be subdivided into 4 classes: 1) endostatin-mediated endocytosis, 2) cellular membrane crypt-like invagination, 3) macroendocytosis, and 4) phagocytosis. Endostatin-mediated endocytosis is mediated by small (about 100nm in diameter) vesicles that have a topographically characterized shell consisting of protein complexes primarily associated with the cytosolic protein endostatin. Thus, in certain embodiments, the endocytic pathway for mitochondrial transfer is an endosomal-dependent endocytic pathway. In other embodiments, the endocytic pathway for mitochondrial transfer is an endostatin-independent pathway. In a specific embodiment, the endocytic pathway is megapinocytosis.
Macropinocytosis has been shown to be an important process in nutrient deprivation environments. Thus, it is hypothesized that cell nutrient deficiency or inhibition by sufficiently nutrient-activated pathways or target molecules (such as mTOR) may be a strategy to enhance cellular engulfment of intact mitochondria to the cytosol. In particular, as provided herein, inhibition of mTOR was found to increase uptake by exogenous mitochondria. mTOR is an essential sensor of amino acids, energy, oxygen, and growth factors, and is a key regulator of protein, lipid, and nucleotide synthesis involved in extracellular nutrient uptake. Thus, in some embodiments, the methods provided herein further comprise contacting the recipient cell with a small compound, peptide, or protein that can increase megakaryocytic drink. In particular embodiments, the methods provided herein further comprise modulating cellular metabolism of the recipient cell prior to transfer of exogenous mitochondrial and/or exogenous mtDNA. In certain embodiments, the regulation of cellular metabolism is performed using the same small compounds, peptides or proteins that can increase macropinocytosis.
Modulation of cellular metabolism can be achieved by any of several well-known techniques, including, but not limited to, those described herein and in the cited references. For example, in some embodiments, the regulation of cellular metabolism is performed by nutrient starvation or nutrient deprivation. In other embodiments, the modulation of cellular metabolism is by a chemical inhibitor or small molecule. In a specific embodiment, the chemical inhibitor or the small molecule is an mTOR inhibitor.
A variety of compounds are known to inhibit mTOR, including rapamycin, also known as sirolimus (CAS number 53123-88-9; C)51H79NO13) And rapamycin derivatives (e.g., rapamycin analogs, also known as "rapalogs"). Rapamycin derivatives include, for example, temsirolimus (CAS number 162635-04-3; C56H87NO16) Everolimus (CAS No. 159351-69-6; c53H83NO14) And bendiolimus (CAS number 572924-54-0; c53H84NO14P). Thus, in some embodiments, the methods for mitochondrial transfer provided herein further comprise modulating cellular metabolism of the recipient cell with rapamycin or a derivative thereof prior to transfer of the exogenous mitochondrial and/or exogenous mtDNA. It is to be understood that the embodiments described above for modulating cellular metabolism are non-limiting, and that modulating cellular metabolism need not include compounds or small molecules.
Thus, in some embodiments, rapamycin or a derivative thereof including a clinically-approved drug can be used as a sole method or in combination with any of the methods provided herein (e.g., a method comprising a reduction in the fraction of endogenous mitochondria of recipient cells) to increase the efficiency of transfer of exogenous mitochondria.
One skilled in the art will appreciate that other delivery methods can also be used to introduce exogenous mitochondrial and/or exogenous mtDNA, and megakaryosis is an exemplary route. In some embodiments, mtDNA can be delivered by endostatin-dependent endocytosis or endostatin-independent endocytosis. In particular embodiments, the endostatin-independent pathway may be, for example, the CLIC/GEEC endocytosis pathway, Arf 6-yi Dependent endocytosis, lipovalve structural protein-dependent endocytosis, macroendocytosis, circular membrane ruffles (circular nuclear ruffles), phagocytosis, or trans-endocytosis. It will also be appreciated that delivery of exogenous mitochondrial and/or exogenous mtDNA can be enhanced by using any compound that stimulates mitochondrial delivery, such as endocytic activators. Non-limiting exemplary compounds suitable for activating endocytosis include, for example, phorbol-12-myristate-13-acetate (PMA) (C)36H56O8) 12-O-tetradecanoyl phorbol 13-acetate (TPA) (C)36H56O8) Tanshinone IIA sodium sulfonate (TSN-SS) (C)19H17O6S.na) and phorbol-12, 13-dibutyrate or a derivative thereof. Furthermore, it will also be understood that methods of non-endocytosis mediated mtDNA and/or mitochondrial transfer, including bypassing endocytosis and/or cell fusion, may be used.
5.4Method of treatment
Provided herein are various methods for treating conditions associated with mutant mtDNA and/or dysfunctional mitochondria, the use of compositions for treating conditions associated with mutant mtDNA and/or dysfunctional mitochondria and the use of compositions in the manufacture of a medicament for treating conditions associated with mutant mtDNA and/or dysfunctional mitochondria. Also provided are methods of treatment comprising restoring or increasing endogenous mitochondrial function using exogenous mitochondrial and/or exogenous mtDNA, restoring or increasing endogenous mitochondrial function using compositions and use of compositions in the manufacture of a medicament for treating a subject in need of mitochondrial replacement. In certain embodiments, the treatment comprises prevention of mitochondrial dysfunction.
5.4.1 methods of treating age-related disorders
In certain embodiments, provided herein are methods of treating a subject having or suspected of having an age-related disease, including any of the methods described in section 5.2 and/or section 5.3. In some embodiments, provided herein are methods of treating a subject having or suspected of having an age-related disease comprising generating a mitochondrial-replaced cell ex vivo or in vitro by: contacting a recipient cell with an agent that reduces endogenous mtDNA or an agent that reduces endogenous mitochondrial function, incubating the recipient cell with the agent for a period of time sufficient to partially reduce mtDNA copy number in the recipient cell or partially reduce the endogenous mitochondrial function, co-incubating (1) a recipient cell in which endogenous mtDNA or endogenous mitochondrial function has been partially reduced and (2) an exogenous mitochondrion from a healthy donor and/or an exogenous mtDNA for a period of time sufficient to non-invasively transfer the exogenous mitochondrion to the recipient cell, thereby producing a mitochondrially replaced cell, and then administering to a subject having or suspected of having an age-related disease a therapeutically effective amount of the mitochondrion replaced recipient cell.
In certain embodiments, the age-related disease comprises an autoimmune disease, a metabolic disease, a genetic disease, cancer, a neurodegenerative disease, and immunosenescence. The metabolic disease may include diabetes. Non-limiting examples of neurodegenerative diseases that can be treated by the methods provided herein include alzheimer's disease or parkinson's disease. In addition, genetic diseases that can be treated include early-aging syndrome, vorner syndrome, and huntington's disease. Other age-related diseases involving dysfunctional mitochondria are also contemplated.
In certain embodiments, the method of treating a subject having or suspected of having an age-related disease comprises producing a MirC, wherein the recipient cell used to produce the MirC is a T cell or a Hematopoietic Stem Cell (HSC). For example, endogenous mtDNA, endogenous mitochondria, or a combination thereof in senescent T cells or Hematopoietic Stem Cells (HSCs) can be replaced for rejuvenation. In vitro or ex vivo mitochondrial replacement may be a viable option for treating patients with disease using human T cells and/or hematopoietic stem cells. Thus, in some embodiments, the methods provided herein can be used to delay senescence and/or extend longevity in a cell by: isolated exogenous mitochondria from healthy, non-senescent cells are non-invasively transferred into senescent or near-senescent cells to rejuvenate recipient cells, and the resulting rejuvenated MirC can then be administered to patients having or suspected of having an age-related disease.
As demonstrated herein, rejuvenation of aging T cells is one possible embodiment by which the invention can be used to treat subjects suffering from age-related diseases, such as cancer. For example, isolated mitochondria from young, healthy T cells autologous to a subject having an age-related disease, such as cancer, for example, can be non-invasively transferred by rejuvenating old T cells that exhibit a senescence-associated secretory phenotype (SASP), including inflammatory cytokines, growth factors, and proteases, reduced and/or slowed rates of cell population doubling, shortened telomeres, increased DNA Damage Response (DDR), or a combination thereof, using the methods provided herein. Then, T cell-derived mircs having characteristics of young, non-senescent cells can be administered to the subject for treatment of age-related diseases.
Thus, in a specific embodiment, a method of treating a subject having or suspected of having an age-related disease comprises the production of MirC, wherein the recipient cell is a T cell. T cell fate is regulated by metabolic pathways, where glycolysis or oxidative phosphorylation (OXPHOS) is responsible for supplying most of the energy to T cells. Glycolytic dominant T cells were selected to differentiate into effector T cells, while OXPHOS dominant T cells were used for memory T cells. Thus, exogenous mitochondrial and/or mtDNA can be used to modulate T cell fate. For example, in the case of allergy, exogenous mitochondrial and/or mtDNA can be used to calm hyperactivated T cells. In other cases, such as in cancer immunotherapy, exogenous mitochondrial and/or mtDNA can energize anti-tumor T cells (empower) to sustain T cells for longer periods of time or to facilitate T cell lytic capacity and/or reduce tumor burden. Furthermore, emerging therapies using chimeric antigen receptor T cells (CAR T) use autologous T cells. Those CAR T cells may be fatigued due to aging or malnutrition, such as cachexia, which is common in the severe pathological stages of cancer. Mitochondrial replacement technology can provide energy and rejuvenate CAR T to provide more ATP, leading to better results.
Thus, in certain embodiments, a method of treating a subject includes recipient cells that are T cells. The T cell may be a CD4+ T cell, a CD8+ T cell, or a CAR T cell. In particular embodiments, mitochondrial replacement in recipient T cells results in T cells with an extended lifespan. For example, the lifetime may be increased by a factor of about 1.5, about 2, about 3, about 4, about 5, or more than 5. In particular embodiments, mitochondrial replacement in the recipient T cell inhibits or delays recipient T cell senescence compared to a T cell without mitochondrial replacement. As described in section 5.2, mtDNA replacement can be performed using exogenous mitochondrial and/or exogenous mtDNA from donor cells younger than recipient cells, thereby extending lifespan. In certain embodiments, the PDL difference between the donor and recipient cells is about 1.5 fold, about 2 fold, about 2.5 fold, about 3 fold, about 4 fold, about 5 fold, or greater than 5 fold. In other embodiments, the donor and recipient cells are from subjects that are about 5 years, about 10 years, about 15 years, about 20 years, or greater than 20 years old apart. In other specific embodiments, mitochondrial replacement in recipient T cells results in T cells with increased cytolytic capacity relative to T cells without mitochondrial replacement. In other embodiments, mitochondrial replacement in T cells results in a decrease in tumor burden.
Although in certain embodiments, plasmid-based gene transfection may be used to generate T cells with exogenous mitochondrial and/or exogenous mtDNA, in other embodiments mRNA transfection may be used. The use of mRNA transfection may reduce the probability of RNA sequences integrating into the host genome and may also have minimal long-term gene expression that would result in a reduction of endogenous mtDNA.
In certain embodiments, the MaxCyte electrotransformer may be used for mRNA transfection, particularly in a clinical setting, which has passed the standards of good manufacturing practice and good clinical practice. Transfection may be performed using a MaxCyte electrotransformation machine, according to the manufacturer's protocol.
Methods of treating a subject having or suspected of having an age-related disease can further comprise the production of mircs using the methods provided herein, wherein the recipient cells are Hematopoietic Stem Cells (HSCs). Hematopoietic Stem Cells (HSCs) provide not only blood cells, but also endothelium that immobilizes damaged resident cells in a distant organ, for example, by transdifferentiation. Furthermore, dysfunction of HSCs has been reported to be involved in systemic senescence. Thus, it has been contemplated that HSC-derived mircs can be used as a treatment in any age-related disease.
In addition, allogeneic HSC transplantation can lead to graft rejection or even graft versus host disease. Autologous HSC transplantation is often a safer and more practical intervention for disease. For example, autologous HSC transplantation typically does not require pretreatment by immunosuppressive agents, such as radiation and chemical agents. Thus, it is contemplated that using the methods provided herein, mircs are produced in vitro or ex vivo using exogenous mtDNA from healthy young mitochondria in autologous HSCs and then returned to the patient's body.
In certain embodiments, the HSC are autologous to the subject in need of mitochondrial and/or mtDNA replacement, and the exogenous mtDNA is allogeneic. As provided herein, mtDNA replacement in HSCs can result in the generation of differentiated cells with functional mitochondria and/or differentiated cells with improved function. Thus, the methods provided herein can be used in the context of HSC transplantation.
Aging alters biological processes and leads to the development of degenerative disorders such as alzheimer's disease, atherosclerosis, osteoporosis, type 2 diabetes, and tissue fibrosis that contributes to chronic kidney disease and chronic obstructive pulmonary disease. Mitochondria can play a role in aging through reactive oxygen species produced by mitochondria, which can affect the aging process. Mitochondrial dysfunction in aging is in a vicious cycle associated with dystrophosis, in which Nicotinamide Adenine Dinucleotide (NAD) is caused by downregulation of nicotinamide phosphoribosyltransferase (NAMPT) and hyperactivation of poly (ADP-ribose) polymerase 1(PARP1) +) The deficiency results in NAD+-inhibition of deacetylase sirtuin 1(SIRT 1). It then goes to acetylation-dependent inactivation of PGC1 α, thus resulting in NAD+A reduction in mitochondrial biogenesis with amplified availability. The low activity of PGC1 α results not only in down-regulation of expression of the encoded mitochondrial protein in the nucleus, but also in down-regulation of expression of the mitochondrial transcription factor TFAM adjacent to the mitochondrial DNA.
In addition to the dual core senescence-regulatory pathway comprising p53 and p16/Rb, the senescence-associated secretory phenotype (SASP), in which a series of inflammatory cytokines, chemokines and proteases, such as IL-1, IL-6/VEGF, IL-8 and CXCL9/MMP, are released, is one of the most characteristic phenomena in senescence. By selective autophagy under normal conditions, the transcription factor GATA4 is degraded by binding to the autophagy linker p62, whereas the DNA Damage Response (DDR) kinases ATM (ataxia telangiectasia mutated) and ATR (ataxia telangiectasia and Rad 3-associated) receive senescence signals to facilitate dissociation between GATA4 and p62 and stabilize GATA4, which in turn activates NF-kB and supports SASP via TRAF3IP2 (tumor necrosis factor receptor-related factor interacting protein 2) and IL 1A. SASP was completely blocked in rho0 cells (mtDNA free cells established by forced mitophagy). In experimental IVF, mitochondrial replacement of oocytes from old subjects ensures promotion of zygote formation, embryonic development and implantation, and success rate of pregnancy.
Impaired protein homeostasis (protein homeostasis) is another characteristic of aging. Protein homeostasis is strictly maintained by translational regulation, protein folding chaperones, ubiquitin-proteasome system (UPS) and autophagy-lysosome system. Since chaperonins are based on ATP, the reduced bioenergy with aging impairs the function of correcting protein folding. Both UPS and autophagy-lysosomal systems, including mitochondrial autophagy, decline over time. The alternation of these 3 systems produces aggregates that are not recirculated in the cytosol, leading to degenerative disorders. In the mitochondrial matrix, accumulation of abnormal proteins will not only initiate the system to degrade it, but will also restore the nuclear communication function of the mitochondria (known as the mitochondrial Unfolded Protein Response (UPR)mt) Provide an opportunity. All of the above pathways include mitochondria. Mitochondrial replacement in somatic cells can disrupt the deleterious aggravating cycle of aging, slow the aging process and even rejuvenate the cells.
Thus, the methods provided herein provide a clinically viable approach to the treatment of heterogeneity and/or to treat various diseases, such as diseases associated with aging, by replacing endogenous dysfunctional mitochondria, such as endogenous mitochondria, with young and/or healthy mitochondria, which may be of autologous or allogeneic origin, using mutant mtDNA.
In some embodiments, the methods for mitochondrial replacement provided herein can be used to treat mitochondrial diseases or disorders as well as aging, cancer, and immune system deficiencies.
5.4.2 methods of treating mitochondrial diseases or disorders
Also provided herein, according to any of the methods described in section 5.2 and/or section 5.3, is a method of treating a subject having or suspected of having a mitochondrial disease or disorder. In some embodiments, a method of treating a subject having or suspected of having a mitochondrial disease or disorder comprises producing MirC according to any of the methods described in section 5.2 and/or section 5.3 and then administering to the subject having or suspected of having a mitochondrial disease or disorder a therapeutically effective amount of a mitochondrially-replaced recipient cell.
A variety of mitochondrial diseases or disorders are known and can all be treated using the methods provided herein. For example, a mitochondrial disease or disorder that can be treated using the methods provided herein can be complex I deficiency (OMIM: 252010). Complex I deficiency is caused by mutations in any of its subunits. In another embodiment, the complex I deficiency is caused by a mutation in a gene selected from the group consisting of: NDUFV1(OMIM:161015), NDUFV2(OMIM:600532), NDUFS1(OMIM:157655), NDUFS2(OMIM:602985), NDUFS3(OMIM:603846), NDUFS4(OMIM:602694), NDUFS6(OMIM:603848), NDUFS7(OMIM:601825), NDUFS8(OMIM:602141), and NDUFA2(OMIM: 602137).
In addition, a mitochondrial disease or disorder that can be treated using the methods provided herein can be complex IV deficiency (cytochrome c oxidase; OMIM: 220110). Complex IV deficiency is caused by mutations in any of its subunits. In certain instances, the complex IV deficiency is caused by a mutation in a gene selected from the group consisting of: MTCO1(OMIM:516030), MTCO2(OMIM:516040), MTCO3(OMIM:516050), COX10(OMIM:602125), COX6B1(OMIM:124089), SCO1(OMIM:603644), FASTKD2(OMIM:612322), and SCO2(OMIM: 604272).
A mitochondrial disease or disorder can be caused by mutation or it can be associated with mutation. The mutations may be point mutations, missense mutations, deletions and insertions. It is understood that the identification of mutations in mtDNA or nDNA is within the skill of one of skill in the art and exemplary methods are provided herein, such as, for example, Single Nucleotide Polymorphism (SNP) assays or droplet digital PCR.
Non-limiting examples of specific types of mitochondrial diseases or disorders that can be treated using the methods provided herein include ornithine transcarbamylase deficiency (OTCD), carnitine O-palmitoyl transferase II deficiency (CPT2), fumarate deficiency, cytochrome c oxidase deficiency associated with Leigh syndrome, Maple Syrup Urine Disease (MSUD), medium chain acyl-coa dehydrogenase deficiency (MCAD), very long chain acyl-coa dehydrogenase deficiency (LCAD), trifunctional protein deficiency, progressive extraocular paralysis with mitochondrial DNA deficiency (POLG), DGUOK, TK2, pyruvate decarboxylase deficiency, and Leigh Syndrome (LS). In another embodiment, the mitochondrial disease or disorder is selected from alpers' disease; a bus syndrome; beta-oxidation defects; carnitine-acyl-carnitine deficiency; carnitine deficiency; coenzyme Q10 deficiency; deficiency of Complex II (OMIM:252011), deficiency of Complex III (OMIM:124000), deficiency of Complex V (OMIM:604273), LHON-Leber hereditary optic neuropathy; MM-mitochondrial myopathy; LIMM-lethal infant mitochondrial myopathy; MMC-maternal myopathy and cardiomyopathy; NARP-neurogenic myasthenia, ataxia and retinitis pigmentosa; leigh disease; FICP-lethal infantile cardiomyopathy and MELAS-related cardiomyopathy; MELAS-mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes; LDYT-Leber hereditary optic neuropathy and dystonia; MERRF-myoclonic epilepsy with ragged red muscle fibers; MHCM-maternally inherited hypertrophic cardiomyopathy; CPEO-chronic progressive external ophthalmoplegia; KSS-Kearns Sayre syndrome; DM-diabetes; DMDF diabetes + deafness; CIPO-chronic intestinal pseudo-obstruction with skeletal muscle degeneration and ophthalmoplegia; DEAF-a DEAFness inherited by the maternal line; PEM-progressive encephalopathy; SNHL-sensorineural hearing loss; encephalomyopathy; mitochondrial cell disease; democho-dementia and chorea; AMDF-ataxia, myoclonus; ESOC epilepsy; optic atrophy; FBSN familial bilateral striatal necrosis; FSGS focal segmental glomerulosclerosis; LIMM lethal infant mitochondrial myopathy; MDM skeletal muscle degeneration and diabetes; MEPR myoclonic epilepsy and psychomotor withdrawal; MERME MERRF/MELAS overlay disease; hypertrophic cardiomyopathy inherited from MHCM maternal lines; cardiomyopathy inherited from the MICM maternal line; leigh syndrome inherited by the MILS maternally; mitochondrial encephalocardiomyopathy; multiple system mitochondrial disorders (skeletal muscle degeneration, encephalopathy, blindness, hearing loss, peripheral neuropathy); NAION non-arteritic inflammatory anterior ischemic optic neuropathy; PEM progressive encephalopathy; PME progressive myoclonic epilepsy; RTT Rett syndrome; sudden infant death syndrome; and MIDD maternally inherited diabetes and deafness.
In particular embodiments, the methods provided herein for treating a mitochondrial disease or disorder can further comprise a mitochondrial disease or disorder caused by an abnormality in mitochondrial DNA, wherein the mitochondrial DNA abnormality is selected from the group consisting of Chronic Progressive External Ophthalmoplegia (CPEO), Pearson Syndrome, Kearn-Sayre Syndrome (KSS), diabetes with deafness (DAD), Leber Hereditary Optic Neuropathy (LHON), LHON-plus (LHON-plus), neuropathy, ataxia and retinitis pigmentosa Syndrome (NARP), Maternally Inherited Leigh Syndrome (MILS), mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS), myoclonic epilepsy with ragged-red fibrosis (MERRF), familial bilateral striatal necrosis/striatal steatosis (FBSN), Luft's disease, aminoglycoside-induced deafness (AID), and various deletions of mitochondrial DNA Syndrome.
Mutations in mtDNA are thought to be associated with a variety of clinical conditions. In adults, these include neurological diseases (e.g., migraine, stroke, epilepsy, dementia, skeletal muscle degeneration, peripheral neuropathy, diplopia, ataxia, speech disorders, and sensorineural deafness), gastrointestinal diseases (e.g., constipation, irritable bowel, and dysphagia), cardiac diseases (e.g., heart failure, cardiac conduction block, and cardiomyopathy), respiratory diseases (e.g., respiratory failure, hypoventilation at night, recurrent aspiration and pneumonia), endocrine diseases (e.g., diabetes, thyroid disease, parathyroid disease, and ovarian failure), ophthalmic diseases (e.g., optic atrophy, cataracts, ophthalmoplegia, and ptosis). In children, disorders believed to be associated with mtDNA mutations include neurological diseases (e.g., epilepsy, skeletal muscle degeneration, psychomotor retardation, ataxia, stiffness, dystonia, and sensorineural deafness), gastrointestinal diseases (e.g., emesis, developmental arrest, and dysphagia), cardiac diseases (e.g., double-chambered hypertrophic cardiomyopathy and dysrhythmias), respiratory diseases (e.g., central hypoventilation and apnea), hematologic diseases (e.g., anemia and pancytopenia), renal diseases (e.g., tubular defects), liver diseases (e.g., liver failure), endocrine diseases (e.g., diabetes and adrenal failure), and ophthalmic diseases (e.g., optic atrophy). Thus, the methods and compositions provided herein are intended for the treatment or prevention of diseases and disorders associated with mutations in mtDNA.
In other specific embodiments, the methods provided herein enable the treatment of a mitochondrial disease or disorder, wherein the mitochondrial disease or disorder is caused by a nuclear DNA abnormality selected from the group consisting of mitochondrial DNA deletion syndrome-4A, mitochondrial recessive ataxia syndrome (MIRAS), mitochondrial neuro-gastrointestinal encephalomyopathy (MNGIE), mitochondrial DNA deletion syndrome (MTDPS), DNA polymerase gamma (POLG) -associated disorders, sensory ataxia-dysregulated neuropathy with dysarthria and ophthalmoplegia (SANDO), leukoencephalopathy with brain stem and spinal cord involvement and elevated lactate (LBSL), coenzyme Q10 deficiency, Leigh syndrome, mitochondrial complex abnormalities, fumarate deficiency, alpha-ketoglutardialdehyde dehydrogenase complex (KGDHC) deficiency, succinyl-coa ligase deficiency, pyruvate dehydrogenase complex deficiency (PDHC)' PDHC Pyruvate Carboxylase Deficiency (PCD), carnitine palmitoyltransferase I (CPT I) deficiency, carnitine palmitoyltransferase II (CPT II) deficiency, carnitine-acyl-carnitine (CACT) deficiency, autosomal dominant-/autosomal recessive-progressive extraocular paralysis (ad-/ar-PEO), infantile spinocerebellar atrophy (IOSCA), Mitochondrial Myopathy (MM), Spinal Muscular Atrophy (SMA), growth arrest, aminouria, cholestasis, iron overload, early death (GRACILE), and Charcot-Marei-Tooth type 2A (CMT 2A).
Many individuals with mtDNA mutations exhibit a group of clinical features belonging to individual clinical syndromes, such as cohn-saiya Syndrome (KSS), chronic progressive extraocular paralysis (CPEO), mitochondrial encephalomyopathy with lactic acidosis and stroke-like attacks (MELAS), myoclonic epilepsy with fragmented red fibers (MERRF), neurogenic weakness with ataxia and retinitis pigmentosa (NARP) or Leigh Syndrome (LS). However, there are a large number of clinical differences and multiple individuals do not match perfectly into a particular class, which is well documented by the overlapping spectrum of disease phenotypes (including the mitochondrial recessive ataxia syndrome (MIRAS) produced by mutations in the nuclear gene POLG, which appears as a major cause of mitochondrial disease or disorder).
Exemplary diseases in which mitochondrial damage is known to play an important role include, but are not limited to, the pathological development of a variety of neurodegenerative diseases, including alzheimer's disease, parkinson's disease, huntington's disease, and amyotrophic lateral sclerosis. In addition, mitochondrial diseases or disorders are subdivided into several syndromes according to the symptoms, not the mutation type. For example, mitochondrial syndromes include mitochondrial myopathy, encephalomyopathy, lactic acidosis, stroke-like symptoms (MELAS), myoclonic epilepsy with fragmented red fibers (MERRF), and Leigh syndrome.
5.4.3 methods of treating a subject in need of mitochondrial replacement
Also provided herein are methods of treating a subject in need of mitochondrial replacement according to any of the methods described in section 5.2 and/or section 5.3. In some embodiments, a method of treating a subject in need of mitochondrial replacement comprises generating a MirC according to any of the methods described in section 5.2 and/or section 5.3 and then administering to the subject in need of mitochondrial replacement a therapeutically effective amount of a mitochondrial-replaced recipient cell.
Subjects in need of mitochondrial replacement include any subject with dysfunctional mitochondria. In certain embodiments, a subject in need of mitochondrial replacement suffers from an age-related disease, a mitochondrial disease or disorder, a neurodegenerative disease, a retinal disease, diabetes, an auditory disorder, a genetic disease, or a combination thereof. Neurodegenerative diseases that may benefit from mitochondrial replacement include, but are not limited to, Amyotrophic Lateral Sclerosis (ALS), huntington's disease, alzheimer's disease, parkinson's disease, friedrich's ataxia, peroneal muscle atrophy, and cerebral leukosis. The retinal disease may be wet or dry age-related macular degeneration, macular edema, or glaucoma. Other exemplary diseases, such as age-related diseases and/or mitochondrial diseases or disorders are described in more detail in sections 5.4.1 and 5.4.2.
Subjects in need of mitochondrial replacement may also include subjects susceptible to mitochondrial dysfunction and asymptomatic. For example, the subject may have mutant mtDNA but no manifestation of mitochondrial disease (for example), and thus the disease is an adult-onset disease. Thus, the methods provided herein can also be used to prevent any of the diseases described herein by treating a subject in need of mitochondrial replacement.
5.5Method for generating iPSC
The present invention also provides methods for producing or increasing the yield of induced pluripotent stem cells (ipscs) from non-pluripotent cells, as described in sections 5.2 and 5.3. Exogenous expression using desiccating factors such as Oct3/4, Klf4, Sox2, and c-Myc has been shown to produce ipscs from non-pluripotent cells. In addition, small copies of mitochondrial DNA (mtDNA) have been detected in undifferentiated ESCs, while this number increases with mitochondrial maturation levels once differentiated (Facucho-Oliveira JM et al, J Cell Sci 2007; 120(Pt 22): 4025-. Thus, the present invention has also identified that the methods provided herein can be used to increase iPSC production by reducing endogenous mtDNA in a non-pluripotent cell by contacting a recipient non-pluripotent cell with an agent that reduces endogenous mtDNA, incubating the recipient non-pluripotent cell with the agent for a sufficient period of time to partially reduce endogenous mtDNA in the non-pluripotent cell, and then introducing one or more expression cassettes for expression of Oct3/4, Klf4, Sox2, and c-Myc. In certain embodiments, the exogenous mtDNA and/or exogenous mitochondria are non-invasively transferred into the recipient cell.
It is understood that the introduction of one or more expression cassettes for expression of Oct3/4, Klf4, Sox2, and c-Myc can occur before, during, or after the introduction of an agent that reduces endogenous mtDNA. Thus, in some embodiments, a method for producing ipscs comprises introducing one or more expression cassettes for expression of Oct3/4, Klf4, Sox2, and c-Myc, contacting a recipient non-pluripotent cell with an agent that reduces endogenous mtDNA and incubating the recipient non-pluripotent cell with the agent for a sufficient period of time to partially reduce endogenous mtDNA in the recipient cell.
In certain embodiments, the method further comprises incubating the recipient cell with the exogenous mitochondrial and/or exogenous mtDNA for a period of time sufficient to non-invasively transfer the exogenous mitochondrial and/or exogenous mtDNA to the recipient cell. In particular embodiments, the method further comprises incubating the recipient cell with the exogenous mitochondria and/or exogenous mtDNA for a period of time sufficient to replace a substantial portion of the endogenous mtDNA. Methods of producing ipscs from non-pluripotent cells comprising transferring exogenous mitochondria and/or exogenous mtDNA and/or exogenous mitochondria can further comprise any of the embodiments described in section 5.3.
Because small copies of mitochondrial dna (mtdna) have been detected in undifferentiated Embryonic Stem Cells (ESCs), the methods provided herein can also be used to promote pluripotency in non-pluripotent stem cells and reduce the exogenous gene factors required to produce ipscs. For example, in some embodiments, the methods provided herein can be used to produce ipscs by reducing endogenous mtDNA in a non-pluripotent cell by contacting a recipient non-pluripotent cell with an agent that reduces endogenous mtDNA, incubating the recipient non-pluripotent cell with the agent for a sufficient period of time to partially reduce endogenous mtDNA in the non-pluripotent cell, and then introducing one or more of Oct3/4, Klf4, Sox2, and c-Myc into the non-pluripotent cell, thereby producing a pluripotent stem cell. In some embodiments, ipscs can be produced even using only small molecule reagents and no exogenous factors.
In certain embodiments, the iPSC contains mutant mtDNA. For example, the mutant mtDNA can contain a point mutation, such as, for example, a point mutation in tRNA (e.g., MELAS). The mutant mtDNA may also include mtDNA having a long deletion of mtDNA. In other embodiments, the non-pluripotent cells for use in generating ipscs are heterogeneous. Incorporation of mutant mtDNA can be beneficial, for example, in the generation of disease models.
In some embodiments, the non-pluripotent receptor cell is a somatic cell. In a specific embodiment, the non-pluripotent cell is a fibroblast.
Culture conditions, identification and establishment of ipscs are within the skill of those in the art. For example, methods include those provided in U.S. patent nos. 8,058,065 and 8,278,104, which are incorporated herein by reference in their entirety.
5.6Assays for measuring heterogeneity
As previously disclosed, mutant mtDNA and/or heterogeneity can lead to dysfunctional mitochondria. Thus, in conjunction with the methods for mtDNA replacement provided herein, assays for evaluating mitochondrial function and/or mtDNA mutation include any assay known to those of skill in the art that can be used to determine or predict the functionality of mitochondrial and/or mtDNA mutations.
For example, assays that determine mitochondrial function include, for example, measurements of any of the following: secreted factors associated with aging (e.g., pro-inflammatory cytokines, proteases, and growth and angiogenesis factors, such as IL-1, IL-6/VEGF, IL-8, and CXCL 9/MMP); mitochondrial function by using orobos; mitochondrial autophagy by use of Keima-red; mitochondrial permeability; mitochondrial membrane potential; cytochrome c levels; active oxygen; respiration of cells; transcriptome and proteomics for measuring activated innate immunity, elimination of hyperactivated glycolysis, alleviation of ER stress, inhibition of the mTOR-S6 pathway and cell cycle activation; mitochondrial dynamics, such as fission and fusion, observed by ultra-precision microscopy and quantified by specialized software; or any assay known in the art that measures mitochondrial function.
A variety of sequencing methods can be used in combination with any of the methods provided herein to (1) detect mutant mtDNA, (2) quantify heterogeneity, and/or (3) assess or confirm metastasis of exogenous mitochondrial and/or exogenous mtDNA. A stretch of approximately 1,100 nucleotides is gene-free and is referred to as the D-loop, and control region. The D-loop contains two regions where mutations accumulate more frequently than anywhere else in the mitochondrial genome. This region is referred to as hypervariable regions HV1 and HV2, respectively. Thus, in some embodiments, mtDNA mutations can be identified by sequencing of the hypervariable region (HV) of the D-loop of mtDNA (i.e., HV1 and/or HV2) in conjunction with the methods provided herein. mtDNA sequencing can be performed using any sequencing method known in the art. In specific embodiments, the sequencing method comprises Single Nucleotide Polymorphism (SNP) determination. In other embodiments, the sequencing method comprises digital PCR. In a specific embodiment, the digital PCR is a droplet digital PCR.
5.7Composition comprising a metal oxide and a metal oxide
Also provided herein are cell compositions obtained by any of the methods described in sections 5.2-5.5. In certain embodiments, provided herein are compositions comprising one or more mitochondria-replaced cells obtained by: (a) contacting a recipient cell with an agent that reduces the copy number of endogenous mtDNA; (b) incubating the recipient cell with the agent for a sufficient period of time to partially reduce the endogenous mtDNA copy number in the recipient cell; and (c) co-incubating (1) the recipient cell from step (b) in which endogenous mtDNA has been partially depleted and (2) exogenous mitochondria for a sufficient period of time to non-invasively transfer the exogenous mitochondria to the recipient cell, thereby producing a mitochondria-replaced cell, wherein the mitochondria-replaced cell comprises greater than 5% exogenous mtDNA. In other aspects, provided herein are compositions comprising one or more mitochondria-replaced cells obtained by: (a) contacting a recipient cell with an agent that reduces the copy number of endogenous mtDNA; (b) incubating the recipient cell with the agent for a sufficient period of time to partially reduce the endogenous mtDNA copy number in the recipient cell; and (c) co-incubating (1) the recipient cell from step (b) in which the endogenous mtDNA has been partially reduced and (2) the exogenous mtDNA from the healthy donor for a period of time sufficient to non-invasively transfer the exogenous mtDNA to the recipient cell, thereby producing a mitochondria-replaced cell, wherein the mitochondria-replaced cell comprises greater than 5% exogenous mtDNA.
The composition can also be obtained by a method comprising contacting a cell with an agent that reduces mitochondrial function and then incubating the recipient cell with the agent for a period of time sufficient to partially reduce endogenous mitochondrial function in the recipient cell. In some embodiments, recipient cells with partially reduced endogenous mitochondrial function can then be co-incubated with exogenous mitochondria from a healthy donor for a sufficient period of time to non-invasively transfer the exogenous mitochondria to the recipient cells, thereby producing mitochondria-replaced cells. In other embodiments, recipient cells with partially reduced endogenous mitochondrial function can then be co-incubated with exogenous mtDNA from a healthy donor for a sufficient period of time to non-invasively transfer exogenous mitochondria to the recipient cells, thereby producing mitochondria-replaced cells. In some embodiments, the mitochondria-replaced cells produced by the methods described above comprise greater than 5% exogenous mtDNA.
As described above, the outer mitochondria can be composed of exogenous mtDNA. Thus, in some embodiments, both exogenous mitochondria and exogenous mtDNA are transferred to the recipient cell and MirC has both exogenous mitochondria and exogenous mtDNA. In other embodiments, the exogenous mtDNA is transferred to the recipient cell by an exogenous mitochondrion and then the exogenous mtDNA is delivered to the endogenous mitochondrion. In particular instances, after delivery of exogenous mtDNA to endogenous mitochondria, the exogenous mitochondria are removed from the cell. Thus, in some embodiments, MirC has exogenous mtDNA, but no exogenous mitochondria.
Since the endogenous mtDNA of the recipient cell is partially degraded, a MirC comprising exogenous mitochondria, exogenous mtDNA, or a combination thereof can contain both exogenous mtDNA and endogenous mtDNA. Similarly, in cases where an exogenous mitochondrion is transferred to a recipient cell, the MirC may contain both exogenous and endogenous mitochondrions. Thus, in particular embodiments, the composition of one or more mitochondria-replaced cells obtained by the methods provided herein has a mixture of endogenous and exogenous mitochondria. In other embodiments, the composition of one or more mitochondria-replaced cells obtained by the methods provided herein has a mixture of endogenous mtDNA and exogenous mtDNA (i.e., heterogeneous mtDNA). In other embodiments, the one or more mitochondria-replaced cells have a total mtDNA copy number of no greater than about 1.1-fold, about 1.2-fold, about 1.3-fold, about 1.4-fold, about 1.5-fold, or more, relative to the total mtDNA copy number of the recipient cell prior to contact with the agent that reduces the endogenous mtDNA copy number.
The invention also includes compositions for use in methods of producing a mitochondrial replaced cell comprising an agent that reduces endogenous mtDNA or an agent that reduces mitochondrial function and a second active agent. In certain embodiments, the composition may further comprise an exogenous mitochondrion, one or more recipient cells, or a combination thereof. In other embodiments, the composition can further comprise exogenous mtDNA.
As described in section 5.3, a plurality of second active agents can be used in a method for producing one or more mitochondria-replaced cells. For example, in some embodiments, the second active agent comprises a macromolecule, a small molecule, or a cell therapy, and the second active agent is optionally selected from rapamycin, NR (nicotinamide ribose), bezafibrate, idebenone, mercaptoethylamine bitartrate (RP103), elamipramide (MTP131), omaviralone (omavelolone) (RTA408), KH176, vatilone (Vatiquinone) (Epi743), lipoic acid, a0001 (alpha-tocopheryl quinone), mitochondrial CoQ10(MitoQ), SkQ1(Visomitin), resveratrol, curcumin, ketonic therapy, hypoxia, and an endocytic activator.
It was found that the use of an endocytic activator increased the uptake of exogenous mitochondria in MTS-XbaIR plasmid treated cells, but had no effect on promoting uptake using "add on" or idle-stained cells, suggesting that this mechanism of exogenous mitochondrial transfer is unique to the invention provided herein. Non-limiting exemplary adapted to activate endocytosisThe compounds include, for example, phorbol-12-myristate-13-acetate (PMA) (C)36H56O8) 12-O-tetradecanoyl phorbol 13-acetate (TPA) (C) 36H56O8) Tanshinone IIA sodium sulfonate (TSN-SS) (C)19H17O6S.na) and phorbol-12, 13-dibutyrate or a derivative thereof. In some embodiments, the endocytic activator comprises a modulator of cellular metabolism.
Modulation of cellular metabolism can be achieved by any of several well-known techniques, including, but not limited to, those described herein and in the cited references. For example, in some embodiments, the regulation of cellular metabolism is performed by nutrient starvation or nutrient deprivation. In other embodiments, the modulation of cellular metabolism is by a chemical inhibitor or small molecule. In a specific embodiment, the chemical inhibitor or the small molecule is an mTOR inhibitor.
A variety of compounds are known to inhibit mTOR, including rapamycin, also known as sirolimus (CAS number 53123-88-9; C)51H79NO13) And rapamycin derivatives (e.g., rapamycin analogs, also known as "rapalogs"). Rapamycin derivatives include, for example, temsirolimus (CAS number 162635-04-3; C56H87NO16) Everolimus (CAS No. 159351-69-6; c53H83NO14) And bendiolimus (CAS number 572924-54-0; c53H84NO14P). Thus, in some embodiments, the compositions provided herein comprise rapamycin or a derivative thereof. It is to be understood that the embodiments described above for modulating cellular metabolism are non-limiting, and that modulating cellular metabolism need not include compounds or small molecules and may include modulation of pathways other than mTOR. It will also be appreciated that the composition may optionally comprise an endocytosis activator and that it is not an essential component. Additionally, in some embodiments, the invention provided herein can include non-endocytosis mediated mtDNA and/or mitochondrial transfer, as in a non-clinical setting.
As described in section 5.5, in certain embodiments, the invention also provides a composition for use in a method of producing an Induced Pluripotent Stem Cell (iPSC) from a non-pluripotent cell, comprising an agent that reduces endogenous mtDNA, one or more expression cassettes for expression of Oct3/4, Klf4, Sox2, and c-Myc, and a recipient cell, wherein the recipient cell is a non-pluripotent cell, wherein the agent that reduces endogenous mtDNA is present in an amount effective to increase the efficiency of producing an Induced Pluripotent Stem Cell (iPSC) from a non-pluripotent cell as compared to a non-pluripotent cell that has not been treated with the agent that reduces endogenous mtDNA. In some embodiments, the agent that reduces endogenous mtDNA is present in an amount effective to increase the efficiency of production of induced pluripotent stem cells (ipscs) from non-pluripotent cells as compared to non-pluripotent cells not treated with the agent that reduces endogenous mtDNA. This is based in part on the following observations: the copy number of mtDNA of pluripotent cells is reduced. In particular embodiments, the composition for use in the method of producing ipscs further comprises exogenous mitochondria and/or exogenous mtDNA.
The invention also includes pharmaceutical compositions for treating an age-related disease, a mitochondrial disease or disorder, a neurodegenerative disease, diabetes, a genetic disease, or any subject in need of mitochondrial replacement, as described in section 5.4. In certain embodiments, provided herein are pharmaceutical compositions comprising an isolated population of cells having mitochondrial replacement of exogenous mitochondria from a healthy donor and cells obtained by the methods described herein, as described in sections 5.2-5.3. In other embodiments, the pharmaceutical composition comprises an isolated population of cells having mitochondrial replacement of exogenous mitochondria and/or exogenous mtDNA from a healthy donor and cells obtained by the methods described herein, as described in sections 5.2-5.3. For example, in some embodiments, a mitochondrially-replaced cell having exogenous mtDNA can optionally further comprise an exogenous mitochondrion. In other embodiments, the exogenous mtDNA is transferred to the cell by the exogenous mitochondria, delivered to the endogenous mitochondria, and then removed from the recipient cell.
The present disclosure also provides an isolated population of cells comprising a mitochondrial replacement with exogenous mitochondria from a healthy donor, wherein the cells are obtained by any of the methods provided herein for obtaining a mitochondrial replacement cell. In another aspect, the present disclosure provides an isolated population of cells comprising a mitochondrial replacement with exogenous mtDNA from a healthy donor, wherein the cells are obtained by any of the methods provided herein for obtaining a mitochondrial replaced cell. In some embodiments, the pharmaceutical composition comprising an isolated population of cells having mitochondrial replacement of exogenous mtDNA from a healthy donor further comprises exogenous mitochondria.
For example, in some embodiments, a pharmaceutical composition comprising exogenous mitochondria from a healthy donor is obtained by a method comprising: contacting a cell with an agent that reduces mtDNA copy number, and then incubating the recipient cell with the agent for a period of time sufficient to partially reduce endogenous mtDNA copy number in the recipient cell. In some embodiments, recipient cells with partially reduced copies of endogenous mtDNA can then be co-incubated with exogenous mitochondria from a healthy donor for a sufficient period of time to non-invasively transfer the exogenous mitochondria to the recipient cells, thereby producing mitochondria-replaced cells. In other embodiments, recipient cells with partially reduced copies of endogenous mtDNA can then be co-incubated with exogenous mtDNA from a healthy donor for a sufficient period of time to non-invasively transfer exogenous mitochondria to the recipient cells, thereby producing mitochondria-replaced cells. In some embodiments, the mitochondria-replaced cells produced by the methods described above comprise greater than 5% exogenous mtDNA.
In other embodiments, the cell is obtained by a method comprising contacting a cell with an agent that reduces mitochondrial function and then incubating the recipient cell with the agent for a period of time sufficient to partially reduce endogenous mitochondrial function in the recipient cell. In some embodiments, recipient cells with partially reduced endogenous mitochondrial function can then be co-incubated with exogenous mitochondria from a healthy donor for a sufficient period of time to non-invasively transfer the exogenous mitochondria to the recipient cells, thereby producing mitochondria-replaced cells. In other embodiments, recipient cells with partially reduced endogenous mitochondrial function can then be co-incubated with exogenous mtDNA from a healthy donor for a sufficient period of time to non-invasively transfer exogenous mitochondria to the recipient cells, thereby producing mitochondria-replaced cells. In some embodiments, the mitochondria-replaced cells produced by the methods described above comprise greater than 5% exogenous mtDNA. The agent that reduces mitochondrial function may reduce mitochondrial function transiently or permanently. It is within the skill of the person of skill in the art to determine the ability of the agent to reduce mitochondrial function either transiently (e.g., reversible inhibitors) or permanently (e.g., irreversible inhibitors).
In certain embodiments of the pharmaceutical compositions provided herein, the cell is obtained by a method further comprising contacting the recipient cell with a second active agent prior to co-incubating the recipient cell with the exogenous mitochondria and/or exogenous mtDNA. In some embodiments, the second active agent is selected from a macromolecule, a small molecule, or a cell therapy, and the second active agent is optionally selected from rapamycin, NR (nicotinamide ribose), bezafibrate, idebenone, mercaptoethylamine bitartrate (RP103), elamipramide (MTP131), omaviralone (omavelolone) (RTA408), KH176, vantiquone (Vatiquinone) (Epi743), lipoic acid, a0001 (alpha-tocopherols), mitochondrial CoQ10(MitoQ), SkQ1(Visomitin), resveratrol, curcumin, a ketonic therapy, hypoxia, and an endocytic activator. In a specific embodiment, the endocytic activator is a modulator of cellular metabolism. In other embodiments, the modulator of cellular metabolism comprises nutrient starvation, a chemical inhibitor, or a small molecule. In other embodiments, the chemical inhibitor or the small molecule is an mTOR inhibitor. In other embodiments, the mTOR inhibitor comprises rapamycin or a derivative thereof.
As described in section 5.2 above, multiple types of cells can be used as recipient cells and donor cells. For example, the present disclosure describes various examples in which the recipient cell is a mammalian cell. However, it will also be understood that any cell having mitochondria can be a recipient cell. Thus, the recipient cell may also be a plant cell.
In some embodiments, the animal cell is a mammalian cell. In a specific embodiment, the cell is a somatic cell. In other embodiments, the somatic cell is an epithelial cell. In other embodiments, the epithelial cells are Thymic Epithelial Cells (TECs).
The present disclosure also provides compositions wherein the somatic cells are immune cells. For example, the composition may comprise an immune cell, wherein the immune cell is a T cell, such as a depleting T cell. In some embodiments, the composition comprises rejuvenated T cells comprising exogenous mitochondria and/or exogenous mtDNA. For example, senescent or near-senescent T cells (e.g., immunosenescence) can be used as recipient cells and T cell-derived mircs can be produced using the methods provided herein to produce T cells with healthy exogenous mitochondria and/or exogenous mtDNA. In a specific embodiment, the T cell is a CD4+ T cell. In other embodiments, the T cell is a CD8+ T cell. In some embodiments, the T cell is a Chimeric Antigen Receptor (CAR) T cell. For example, in some embodiments, the present disclosure provides MirC as CAR-T cells that are effective at killing cancer cells. MirC-derived CART can have prolonged survival to enable improved immune surveillance and improved cancer cell killing. In other embodiments, the immune cell is a phagocytic cell.
As noted above, the compositions provided herein may also include compositions for delaying senescence and/or extending longevity in a cell. The composition can include a senescent or near-senescent cell with endogenous mitochondria, an isolated exogenous mitochondria from a non-senescent cell, and an agent that reduces the copy number of endogenous mtDNA. The composition may also include a senescent or near-senescent cell with endogenous mitochondria, isolated exogenous mitochondria from a non-senescent cell, and an agent that reduces mitochondrial function.
Also provided herein are compositions comprising cells derived from one or more mitochondrial replacements of recipient cells that are bone marrow cells. In specific embodiments, the bone marrow cells are Hematopoietic Stem Cells (HSCs) or Mesenchymal Stem Cells (MSCs). For example, HSCs or MSCs may be isolated from a subject having or suspected of having a mitochondrial disease, an age-related disease, or otherwise in need of mitochondrial replacement, and have endogenous mitochondria replaced with exogenous mitochondria. Subsequently, HSC or MSC derived mircs can then be transplanted back into subjects in need of mitochondrial replacement. In other embodiments, the recipient cell is an iPS cell. The compositions can be used in a clinical setting and can be effective in treating age-related diseases, treating mitochondrial diseases or disorders, treating neurodegenerative diseases, treating diabetes or genetic diseases. For example, in some embodiments, the ipscs can be differentiated into specific cell types prior to administration back to the subject using methods known in the art.
In other embodiments, provided herein is a pharmaceutical composition comprising an isolated population of pluripotent cells with reduced amount of endogenous mtDNA, wherein the cells are obtained by any of the embodiments described in section 5.5. In a specific embodiment, the isolated population of pluripotent cells is iPS cells.
Administration of the cells or compounds described herein is by any route commonly used for the introduction of drugs. The pharmaceutical compositions of the present invention may comprise a pharmaceutically acceptable carrier. In particular embodiments, the term "pharmaceutically acceptable" means approved by a regulatory agency of the federal or a state government or listed in the U.S. pharmacopeia or other generally recognized foreign pharmacopeia for use in animals, and more particularly in humans. The term "carrier" refers to a diluent, adjuvant, excipient, or vehicle with which the therapeutic agent is administered. The pharmaceutically acceptable carrier is determined in part by the particular composition being administered and by the particular method used to administer the composition. Thus, there are a variety of suitable formulations of the Pharmaceutical compositions of the present invention (see, e.g., Remington's Pharmaceutical Sciences, 17 th edition, 1985).
Formulations suitable for administration include aqueous and non-aqueous solutions, isotonic sterile solutions, which may contain antioxidants, buffers, bacteriostats, and solutes that render the formulation isotonic, and aqueous and non-aqueous sterile suspensions that may include suspending agents, solubilizers, thickening agents, stabilizers, and preservatives. In the practice of the present invention, the composition may be administered, for example, orally, nasally, topically, intravenously, intraperitoneally, intrathecally, or into the eye (e.g., by eye drops or injection). The formulations of the compounds may be presented in unit-dose or multi-dose sealed containers, such as ampules and vials. Solutions and suspensions may be prepared from sterile powders, granules and tablets of the kind previously described.
In the context of the present invention, the dose administered to a patient should be sufficient to elicit a beneficial response in said subject over time, i.e. to prevent, ameliorate or improve the condition in the subject. The optimal dosage level for any patient will depend upon a variety of factors including the potency of the particular modulator used, the age, weight, physical activity and dietary profile of the patient and on the possible combinations with other drugs. The size of the dose will also be determined by the presence, nature and extent of any adverse side effects that accompany the administration of a particular compound or carrier in a particular subject. Administration can be accomplished by single or divided doses.
The scope of the invention is not limited by the specific embodiments described herein. Indeed, various modifications of the invention in addition to those described will become apparent to those skilled in the art from the foregoing description and accompanying drawings. Such modifications are intended to be within the scope of the appended claims.
All patents, patent applications, published patent applications and other publications cited herein are incorporated by reference in their entirety. In the event that any description of the term conflicts with any document incorporated by reference, the term description set forth herein shall govern.
Throughout this application, various publications have been referenced. The disclosures of these publications in their entireties are hereby incorporated by reference into this application in order to more fully describe the state of the art to which this invention pertains. While the invention has been described with reference to the examples provided above, it will be appreciated that various changes can be made without departing from the spirit of the invention.
1. Examples of the embodiments
Embodiments are provided in this section by way of illustration and not by way of limitation. The following examples are provided as exemplary embodiments of the present invention. They should not be viewed as limiting the broad scope of the invention.
Example I: optimization of the MirC protocol shows that XbaI degrades mtDNA and MTS expression vector-targeted mitochondria in vitro
Figure 1A provides a scheme for a method for generating a mitochondrial-replaced cell (MirC). First, a mammalian expression vector for expression of the XbaI restriction enzyme fused to the mitochondrial-targeting sequence (MTS) was engineered by cloning the MTS-XbaI sequence into the pCAGGS vector using standard techniques known in the art (FIG. 1B). Among the Mitochondrial Transfer Signals (MTS) reported, we used the ND4 signal sequence in this study. The resulting expression vector also contained a puromycin resistance gene to allow selection (FIG. 1B).
XbaIR is the most powerful endonuclease and in the human mitochondrial genome, the mtDNA standard sequence named under the Cambridge Reference Sequence (CRS) has up to 5 recognition sites targeted by a particular endonuclease (fig. 1D). Isolated mtDNA was hydrolyzed at multiple sites by XbaIR as verified by in vitro endonuclease co-incubation (fig. 1C). In contrast, NotI digestion of mtDNA revealed a single fragment, as predicted by the Cambridge Reference Sequence (CRS) of mitochondrial DNA (fig. 1C).
The gene transfer protocol of plasmid DNA into cells was optimized using Normal Human Dermal Fibroblasts (NHDF) expressing Enhanced Green Fluorescent Protein (EGFP) by using the Nucleofector electroporation-based transfection method. Greater than 90% efficacy and greater than 90% viability were established after 1 day of 2 μ g/ml puromycin exposure (figure 1E).
To specifically evaluate the effectiveness of MTS targeting sequences, a plasmid with MTS fused to EGFP was generated by subcloning the EGFP gene in place of the XbaIR gene to generate pCAGGS-MTS-EGFP-PuroR plasmid (fig. 1F). Then, Normal Human Dermal Fibroblasts (NHDF) were transfected with MTS-EGFP expression vector and counterstained with TMRM (tetramethylrhodamine, methyl ester), a cell-penetrating dye that accumulates in active mitochondria with intact membrane potential (fig. 1G).
Overall, these results indicate that XbaI can be used to digest mitochondrial DNA, cells can be transfected efficiently without affecting cell viability, and that expression vectors containing MTS can efficiently target mitochondria.
Example II: endonuclease MTS-XbaIR treatment showed improved degradation of mtDNA relative to the conventional method of EtBr
The efficiency and efficacy of the MTS-XbaIR expression vector was evaluated relative to the conventional method using ethidium bromide (EtBr) according to the protocol shown in FIG. 2A. Placental venous endothelial-derived cell line EPC100 with DsRed-labeled mitochondria was cultured in pyruvate-free DMEM (Wako cat #044-29765) with 10% Fetal Bovine Serum (FBS) and 1% penicillin/streptomycin (P/S), and on day 0, the cells were untreated ("normal"), transfected with MTS-XbaIR expression vector ("MTS-XbaIR") or treated with 50ng/mL EtBr. On day 1, cells were cultured in DMEM with 10% FBS and 1% P/S supplemented with 100. mu.g/mL pyruvate and 50. mu.g/mL uridine. Quantitative polymerase chain reaction (qPCR) was performed at days 3 and 5 to measure mtDNA relative to housekeeping gene β -actin (Actb) according to methods known in the art. The results show that XbaIR reduced mtDNA copy number to 2715.8141, whereas EtBr treatment only reduced mtDNA copy number to 5169.1258, similar to the DNA copy number of 6189.6867 in untreated cells (fig. 2B). Although the reduction of mtDNA in the endonuclease-treated group was superior to the group treated with the conventional method, it was not completely deleted and about 30% of the endogenous mtDNA remained (fig. 2B). Cells with such a partial reduction in mtDNA are referred to as A cell.
The increased degradation of endogenous mtDNA in the MTS-XbaIR treated group relative to the EtBr group was further confirmed by microscopy of DsRed-labeled mitochondria (fig. 2C). The reduction level was reflected in the remaining healthy mitochondrial volume estimated by TMRM staining, and was lower in the XbaIR-treated group than in the conventional method group (fig. 2C). In addition, FACS analysis of NHDF cells showed a decrease in TMRM after treatment with XbaIR (fig. 2D).
The kinetics of expression of XbaIR after plasmid gene transfer was examined by qPCR. On day 3, expression peaked and then fell to 0 on day 7 (fig. 2E). Other genes of interest (e.g., GFP) demonstrated the same kinetics as XbaIR (fig. 2E). Fluorescence images confirmed GFP enrichment in cells transfected with the MTS-EGFP-PuroR plasmid after puromycin selection compared to before transfection (fig. 2F and 2G). The fraction of GFP positive cells was significantly increased to almost 100% by 1 day exposure to puromycin (fig. 2F).
These results demonstrate that the reduction in mtDNA copy number in the XbaI endonuclease treated group is superior to the group treated with the conventional EtBr method and that the complete deletion of the entire endogenous mtDNA is not complete. Furthermore, transient selection using puromycin enables significant enrichment of cells expressing the MTS construct.
Example III: partial degradation of endogenous mitochondria using MTS-XbaIR constructs in recipient cells enables mitochondrial replacement from exogenous donor cells
To assess whether exogenous mitochondria from healthy donor cells could be transferred to recipient cells with XbaI-mediated mtDNA elimination, NHDF cells were transfected with MTS-GFP or MTS-XbaIR plasmids and selected using puromycin after 48 hours. Isolated mitochondria from a human cell line derived from uterine endothelium labeled with DsRed (termed EPC100) were transferred to donor cells 6 days after transfection. Figure 3A shows a protocol.
Mitochondrial content was assessed by TMRM staining after transfection with MTS-GFP or MTS-XbaI and selection with puromycin. As shown in fig. 3B, MTS-GFP transfected cells showed strong staining for TMRM, indicating high mitochondrial levels in NHDF cells. In contrast, MTS-XbaI transfected cells (p-) showed a decrease in mitochondrial volume as shown by TMRM staining (FIG. 3B).
The reduction of mitochondrial DNA was further confirmed by quantifying the number of mitochondrial DNA by qPCR for 12S-rRNA after nuclear regulation with β -actin (Actb) (fig. 3C). On day 6, mitochondrial DNA from MTS-XbaI transfected cells was significantly reduced (ρ -) (fig. 3C) relative to NHDF control cells transfected with MTS-GFP. The significant reduction of mitochondrial DNA in rho-cells continued throughout the assay period, which ceased on day 12. Specifically, the copy number was reduced to about 1/3 of the original copy number on day 6 and further reduced to about 1/4 on day 12 in ρ -cells (fig. 3C).
Mitochondria were isolated from DsRed-Mt EMC by differential centrifugation. Briefly, a homogenate buffer containing a protease inhibitor cocktail (Sigma-Aldrich, st. louis, Missouri, USA) [ HB; 20mM HEPES-KOH (pH 7.4), 220mM mannitol and 70mM sucrose]Cells were harvested from the culture dish. The cell particles were resuspended in HB and incubated on ice for 5 min. The cells were disrupted on ice by 10 strokes (strokes) of a 27-gauge needle. The homogenate was centrifuged (400g, 4 ℃, 5min) twice to remove unbroken cells. Mitochondria were harvested by centrifugation (6000g, 4 ℃, 5min) and resuspended in HB. The amount of mitochondria isolated was expressed as protein concentration using the Bio-Rad protein assay kit (Bio-Rad, Richmond, Calif., USA). Mitochondrial transfer was performed by CO-incubating the isolated mitochondria with cells in 2ml of standard medium at 37 ℃ for 24h with 5% CO 2. Importantly, isolated mitochondria andco-incubation of cells at day 12 resulted in a significant increase in mtDNA copy number, similar to the levels of control NHDF cells (fig. 3C).
Consistent with the results shown in FIGS. 2C and 2D, after MTS-XbaI transfection,the cells showed a decrease in mitochondrial content as measured by TMRM imaging. Importantly, by mixing Contacting the cells with isolated exogenous mitochondria can rescue the reduction of mitochondria as indicated by uptake of isolated mitochondria labeled by DsRed (fig. 3D and 3E). In contrast, DCo-culture of sRed-labeled and isolated mitochondria with NHDF control cells or NHDF cells transfected with the idling chromosome MTS-EGFP expression vector showed that exogenous mitochondria aggregated around the cells and formed aggregates, but were not internalized (fig. 3D, bottom panel). Although a small fraction of the mitochondria are engulfed, most of them remain extracellular with intact endogenous mitochondria, and the DsRed intensity is maintained during this period. DsRed aggregates become smaller and less, and DsRed intensity decreases, indicating that after foreign mitochondria accumulate on the cell membrane, they are engulfed and their mitochondrial membrane fraction is rapidly digested.
Comparison between the existing methods shows that the endonuclease method of the present invention is more effective in generating cells with mitochondrial replacement of exogenous mitochondria (fig. 3F). For example, the endonuclease method of the present invention is combined with (1) the addition of a mitochondrial transfer method, as described in our previous work (see, e.g., Kitani, t. et al, J Cell Mol Med (2014)18,1694) or (2) the recently reported method of gyrobacterial inoculation (fig. 3F) using isolated mitochondria by metabolizing healthy cells (see, e.g., Kim, m.j. et al, Sci Rep 8,3330, (2018)). None of the previously reported methods (i.e., mitochondrial addition; "Mt addition" or inoculation with 800 xg or 1500 xg gyrobacteria) showed any significant exogenous mitochondrial transfer as measured by FACS analysis of DsRed labeled exogenous mitochondria (fig. 3F). On the other hand, the novel methods provided herein for non-invasive transfer of exogenous mitochondria (Mt EPC100) followed by partial degradation of endogenous mitochondria mediated using MTS-XbaI showed a significant DsRed positive fraction and increased mean fluorescence intensity after exogenous mitochondrial transfer (fig. 3F, top right panel, far right line).
Previously established methods used in mitochondrial biology use cells with complete mitochondrial depletion, cells (see, e.g., filed 9/23 2010 and as US 2011-U.S. patent application No.12/747,771, published as 8 a1, incorporated herein by reference in its entirety). However,the cells were unable to engulf the outer mitochondria (fig. 3G-fig. 3I). Based on the results provided herein, it is assumed thatCells cannot engulf foreign mitochondria due to the energy shortage necessary to carry out macropinocytosis. To confirm this hypothesis, we designed genetically modified cells to be produced by exposure to antimycin that causes mitophagyCells, and mitochondrial transition levels were examined. The results indicate that no external mitochondrial engulfment occurred in cells with complete mitochondrial depletion (fig. 3G-fig. 3I). Thus, these results indicate that partial, rather than complete, deletion of pre-existing mtDNA is a key factor in exogenous and extracellular mitochondrial megapinocytosis.
In addition, uptake of DsRed-labeled exogenous mitochondria was monitored in rho (-) cells treated with or without exogenous mitochondria, untransfected cells (added Mt) or cells treated with empty MTS-GFP plasmid. Fluorescence intensity of DsRed was quantified every 24 hours using NIH imaging software. The relative values for the initial intensity are shown in the histogram (fig. 3J). Quantification indicated that simple addition of mitochondrial co-incubation and mitochondrial co-incubation with empty-transfectants increased intensity to the same extent due to aggregation of isolated mitochondria, indicating accumulation of Ds-red labeled mitochondria rather than engulfment. In contrast, the intensity of rho (-) cells co-incubated with isolated exogenous mitochondria gradually decreased over time, indicating that engulfed mitochondria were degraded.
These results indicate that MTS-XbaI expression vector can produce mitochondria which have been partially deleted for endogenous mitochondriaA cell, and canMitochondrial content is saved by transferring isolated exogenous mitochondria from donor cells. As described herein, the methods of the invention provide improved mitochondrial transfer efficiency relative to previously described methods, such as those performed in conjunction with centrifugation or simple "addition" of mitochondria without partial reduction of endogenous mtDNA. However, cells that are not able to completely degrade in endogenous mitochondria: (Cells), indicating that energy may be required for the uptake by exogenous mitochondria.
Example IV: isolated exogenous mitochondria fused to endogenous mitochondria to transfer donor mtDNA
To further elucidate how mitochondrial transfer of intact mitochondria occurs, the fate of mitochondria transferred into cells was studied separately for the outer and inner membranes and for the mimotopes. In some cases, transient inter-mitochondrial fusion events have been observed in which two mitochondria are closely juxtaposed, exchanging soluble inter-membrane lumen and matrix proteins and re-separating, thereby retaining the original morphology (see, e.g., Liu X et al, EMBO J. 2009; 28(20): 3074-. Thus, transient inter-mitochondrial fusion events were analyzed under the conditions described herein.
Isolated mitochondria from EPC100 donor cells were labeled with DsRed, and recipient cells with EGFP-labeled mitochondria were used. Fig. 4A shows a chart of the protocol used. Microscopic images of transient contact of donor and indigenous mitochondria revealed that no extensive mitochondrial fusion was observed (fig. 4B and 4C). Most donor mitochondria exist independently of endogenous mitochondria. In addition, a small transient fusion image was observed, and then the donor mitochondria appeared to leave before it finally disappeared (fig. 4C).
Mitochondrial transfer was performed according to the protocol shown in fig. 4F. Briefly, mitochondria from recipient NHDF cells were labeled with DsRed-label (fig. 4D) and mitochondria from donor EPC100 cells were labeled with TFAM, which binds to mtDNA and allows for mitochondrial tracing (fig. 4E). Recipient NHDF cells were transfected with pCAGGS-MTS-XbaIR-P2A-Puror expression vector and selected with puromycin on day 2 for 24 hours. On day 6, mitochondrial transfer of TFAM-GFP-labeled mitochondria from EPC100 donor cells was performed. Then, on day 8, cells were imaged. Microscopy of mitochondrial transfer showed migration of the donor pseudonuclear into the pre-existing mitochondrial matrix (fig. 4G). Exogenous mitochondria transiently contact the receptor mitochondria, indicating transfer of mitochondrial nucleomimetics including TFAM to pre-existing mitochondria by transient contact.
These results indicate that the donor mitochondria are transferred to the mitochondrial matrix in the recipient cell and predominate in cases of pre-existing mitochondrial depletion. Furthermore, according to these experiments, almost all isolated mitochondria were engulfed. On the other hand, the addition of mitochondrial transfer and empty-transfectants did not show strict engulfment, but rather a large proportion of these exogenous mitochondria accumulated on the cell surface.
In summary, the results of examples III and IV indicate that ρ (-) cells degraded and engulfed mitochondria (fig. 3J) and were in transient contact with pre-existing mitochondria (fig. 4B-4C), while exogenous mtDNA with TFAM was present in pre-existing mitochondria (fig. 4G).
Thus, it is hypothesized that the outer mitochondria are able to temporarily interact with the inner mitochondria and transport mtDNA during the temporary interaction. The exogenous mitochondrial membrane complex can then be degraded in the cytosol to provide a building block for reconstructing mitochondria. Mitochondria of recipient cells that receive exogenous mitochondria can gradually reconstitute mitochondrial membrane complexes and achieve functional recovery.
Example V: SNP assays detect an increase in exogenous mitochondria following transfer of isolated exogenous mitochondria
To assess the source of mtDNA after mtDNA replacement, different nucleotides identified between NHDF and EPC100 by sequencing hypervariable regions 1 and 2 were used (fig. 5A and 5B). Although NHDF was conserved as a at position 16362 in CRS, EPC100 had a mutation at the same position that resulted in an alteration from a to G (fig. 5B). Importantly, mitochondrial replacementCellsThe evaluation of (a) indicates the presence of both the original nucleotide in the secondary wave and the exogenous nucleotide G in the primary wave, indicating that the cell is heterogeneous (fig. 5B, bottom panel).
Heterogeneity in mitochondrially replaced NHDF was further assessed by single nucleotide polymorphism assays to detect differences between recipient NHDF and donor EPC100 (fig. 5C). The HV1 region was amplified using the hmt16318-F primer (5'-agccatttaccgtacatagcacatt-3' (SEQ ID NO: 6)) and the hmt16414-R primer (5'-cacggaggatggtggtcaag-3' (SEQ ID NO: 9)), and SNPs were detected using the NHDF-specific probe (5'-CTTCTCGTCCCCATG-3' (SEQ ID NO: 5)) and the EPC 100-specific probe (5'-CCCTTCTCGCCCCCAT-3' (SEQ ID NO: 7)) (FIG. 5C). SNP assay results showed that the ratio of EPC100 relative to NHDF reached 66.6% on day 12 after mtDNA substitution (fig. 5D). This result is an unexpected improvement over previous approaches that resulted in human endometrial gland-derived mesenchymal cells engulfming a relatively small fraction of the extracellular mitochondria and having little impact on the level of heterogeneity (see, e.g., Kitani, t. et al, Journal of Cellular and Molecular Medicine,18, 1694-.
These results indicate that the methods provided herein for replacing mtDNA by exogenous mitochondria and/or exogenous mtDNA are entirely novel and an improvement over the prior art. As described herein, the methods provided indicate that mitochondrial translocation can result in exogenous mtDNA becoming the primary mtDNA following MTS-XbaI mediated endogenous mitochondrial degradation.
Example VI: the replaced mitochondria produced energy and the MirC showed a phenotypic recovery similar to that of normal control cells
Whether the alternative mitochondria worked to produce energy was investigated by using orobos O2k according to the manufacturer's instructions. Use of native control cellsThe cells replaced by cells and mitochondria produced representativesThe sexual oxygen consumption rate curve, then the respiratory flow and control ratio were calculated (fig. 6A and 6B). Basal respiration, maximal capacity of the electronic delivery system, and ATP production (Free Routine Activity) all show similar kinetics and suggest that these indices pass throughCells were significantly reduced (fig. 6B, top column). Importantly, these indices returned to the original values by the mitochondria-replaced cells (fig. 6A and 6B). In thatIn cells, non-mitochondrial ATP production (ROX) was up-regulated and the coupling rate was down-regulated (fig. 6B, bottom panel). The energy supply mechanism in the cell is biased towards glycolysis from mitochondrial ATP production, and these changes are reversed after replacement with the native cell's mtDNA (fig. 6B, top right).
In addition, the phenotypic recovery of mitochondrial replaced cells (MirC) was confirmed by their proliferative capacity. In particular, the amount of the solvent to be used,cells showed poor proliferative capacity, while MirC recovered to levels close to control cells on days 6-12 (fig. 6C, right panel).
These results indicate that the method provides mtDNA replacement using clinically useful materials and results in cells with functional mitochondria that enable phenotypic recovery of mitochondrial replaced cells (MirC).
Example VII: mTOR inhibition by rapamycinMegalocytosis of exogenous mitochondria in cells
To determine the method for improving the ability of cells to carry out MirC, the mechanism of regulation of exocytosis of the outer source mitochondria was studied. Because of the fact thatCells are depleted of ATP due to mitochondrial depletion, so it is hypothesized thatThe intracellular high energy state of the cell is similar to the starvation state. To this end, two molecular pathways were investigated: mammalian targets of rapamycin complex 1(mTORC1) and AMP-activated protein kinase (AMPK). mTORC1 is an essential sensor of amino acids, energy, oxygen, and growth factors, and is a key regulator of protein, lipid, and nucleotide synthesis. AMPK is a sensor of AMP levels, and activation leads to autophagy, mitochondrial biogenesis, glycolysis, and lipolysis. Two pathways are involved in the absorption of extracellular nutrients.
As shown in fig. 6D, for the purpose of studyThe mechanism of megalocytosis in cells, AMPK/mTORC1 was stimulated using starvation, while the "drugs" palmitic acid and rapamycin were used to specifically stimulate mTORC1 activation and inhibit mTORC1, respectively. Rapamycin was added to the medium at a concentration of 50ng/ml for 24 hours and the cells were exposed to serum-free medium without glucose and essential amino acids for 1 hour to simulate starvation. While Palmitic Acid (PA) was reported to activate mTORC1 in vivo at a concentration of 200 μ M, for cultured fibroblasts, titration of PA showed that a concentration of 50 μ M and a duration of 24 hours were optimal based on cell viability. By using capillary electrophoresis, WesTM(Protein Simple) ratios of phosphorylated AMPK to AMPK and phosphorylated p 70S 6 kinase to p 70S 6 kinase were examined and are downstream targets of mTORC 1.
Treatment with PA or rapamycin showed thatAMPK pathway was not significantly activated in cells (fig. 6G and 6H), but at levels similar to starvation and rapamycinThe mTORC1 pathway was significantly inhibited in the cells as measured by pS6/S6 (fig. 6E-6F). These results indicate that mTORC1 representsAn important target for mitochondrial macropinocytosis in cells.
Then, during mitochondrial co-culture, we examined the effect of rapamycin and palmitic acid on mitochondrial engulfment by treating cells with rapamycin or palmitic acid simultaneously. Fig. 6I shows the protocol. Briefly, NHDF acceptor cells were transfected with MTS-XbaI expression vector and cultured with or without rapamycin or with or without Palmitic Acid (PA). For expression of MTS-XbaI after 48 hoursCells were subjected to puromycin selection. On day 6, transfer of isolated mitochondria labeled with DsRed from EPC100 cells was performed. On day 8, FACS analysis was performed to detect donor mitochondria by measuring DsRed expression in NHDF acceptor cells.
As shown in fig. 6I-6L, rapamycin treatment significantly improved the engulfment of DsRed-labeled isolated exogenous mitochondria, while palmitic acid significantly inhibited it. These experiments were repeated 4 times and the positive fractions were summarized, which indicates that the pairs in rapamycin and palmitic acidThere were statistically significant differences in the cells (fig. 6I and 6K). Notably, there was no significant difference in both null transfection and additive-type mitochondrial transfer. In addition, the results show that the effect of modulating mTORC1 activity only affects Mitochondrial transfer of cells without affecting "additive" or idle contaminating cells, suggesting that this mechanism of transferring exogenous mitochondria is unique to the invention provided herein.
These results indicate that activation of mTORC1 by rapamycin during mitochondrial transit can increase mitochondrial megakaryosis. Furthermore, these methods demonstrate that rapamycin, a clinically useful drug, can be used to increase megalocytic efficiency for the production of MirC.
Example VIII: mtDNA replacement with heterogeneity Reversal in fibroblasts derived from patients with Leigh syndrome
To investigate whether mitochondrial diseased cells could be corrected by using in vitro mtDNA replacement techniques, primary fibroblasts (7SP) derived from patients diagnosed with Leigh syndrome with the mtDNA T10158C mutation were used as recipient cells (fig. 7A). The same procedure previously described in NHDF cells was applied to 7SP fibroblasts. In EPC100 donor mitochondria, DNA sequencing of mtDNA at 10158 nucleotides verified as T (fig. 7B, top), whereas 7SP fibroblasts had a mosaic of T in the dominant wave and C in the minor wave, indicating heterogeneity (fig. 7B, bottom).
The kinetics of mtDNA content in 7S fibroblasts after mitochondrial replacement was almost identical to that in NHDF (fig. 7C and 7J). Time-delay observation displayFibroblast cells exhibit the sameThe cells behave the same. Specifically, exogenous mitochondria are present inThe aggregates that accumulate on the cell surface become smaller or less over time and the intensity of DsRed in the cytosol decreases rapidly, indicating efficient engulfment into and digestion in the cytosol, in combination with the useThe results produced by the cells were consistent.
Importantly, mtDNA copy number recovered to the same value as the original 7S fibroblasts at day 12 after mitochondrial replacement (fig. 7D). On the other hand, despite the same co-culture with isolated mitochondria under the same conditions, the free-standing chromosome of 7SP fibroblasts (added mitochondrial transfer) did not even increase mtDNA copy number, indicating poor external mitochondrial transfer (fig. 7D, light grey bar).
Mitochondria in 7S fibroblasts were examined for the presence of foreign and healthy mtDNA by sequencing the mitochondrial genome segment containing 10158 nucleotides. As shown in FIG. 7E, following mitochondrial replacement, at receptor 7SPIn the cells, the mtDNA sequence of 7SP cells changed from having most of the mutant heterogeneity (C of large wave and T of small wave) at the 10158 nucleotide position to most of the wild type mtDNA (T of large wave and C of small wave) (fig. 7E, bottom).
To generate quantitative information, Single Nucleotide Polymorphism (SNP) assays are performed to estimate the heterogeneity generated using this technique. The ND3 region of mitochondrial DNA was amplified using hmt10085-F primer (5'-CAACACCCTCCTAGCCTTACTACTAA-3' (SEQ ID NO: 17)) and hmt10184-R primer (5'-GTCGAAGCCGCACTCGTA-3' (SEQ ID NO: 20)) and EPC 100-specific probe (5'-ACATAGAAAAATCCACCC-3' (SEQ ID NO: 18)) or 7 SP-specific probe (5'-CTACATAGAAAAATCCAC-3' (SEQ ID NO: 19)) (FIG. 7F). The results indicated that the level of original hmt10158 heterogeneity in 7SP fibroblasts was about 90% of mutant mtDNA (fig. 7G). At day 12 post-replacement, heterogeneity in 7SP cells receiving mitochondrial metastasis showed a level of heterogeneity as little as 10% (fig. 7G), while the free-standing dye (added mitochondrial metastasis) did not significantly alter heterogeneity and maintained nearly the same ratio or greater than 90% (fig. 7H and 7I). These results indicate that the mitochondrial replacement techniques provided herein are superior to the additive mitochondrial transfer previously reported. Subjected to endonuclease treatmentThe cells improved heterogeneity to about 75%, with mtDNA copy number reduced by about 80%.
Overall, these results indicate that the methods described herein for generating mitochondrial replacement cells using MTS-XbaI to partially reduce endogenous mitochondria can be effectively used in cells from subjects with mitochondrial diseases or disorders to improve the level of heterogeneity and reduce the amount of mutant mtDNA.
Example IX: mtDNA replacement in fibroblasts from patients with Leigh syndrome leads to improved cell life and cell metabolism
The functional activity of mitochondrially replaced 7SP fibroblasts was evaluated. As shown in FIGS. 8A and 8B, at about day 12, mitochondria replaced 7SP fibroblastsCan be restored to levels equivalent to the original 7SP fibroblasts.
In addition, mitochondrially replaced 7SP fibroblastsA significant life extension was shown up to about 63 rd Population Doubling Level (PDL), while doubling time exceeded 120 hours, which is the threshold for growth arrest (fig. 8C). Cells receiving mtDNA replacement at about the 8 th PDL and recombinant cells with healthy mtDNA were able to continue dividing beyond the 55 th PDL, which is considered to be the number of times a normal human cell population will divide before cell division ceases (i.e., the hflik limit). In contrast, untreated 7S fibroblasts entered senescence at the 25 th PDL (fig. 8C). Thus, experiments indicate that mtDNA replacement has a significant impact on the proliferation and longevity of mitochondrial diseased cells. Given that increased aging with aging and cancer cells often includes mitochondrial dysfunction, this approach may provide an important cue for rejuvenation, and this may provide the basis for novel strategies for cancer therapy as well as therapy of other age-related diseases.
The functional role of mitochondrial metastasis in 7S fibroblasts was further evaluated by measuring cell size (fig. 8D). Mutation of 7S fibroblasts in the coding sequence of the ND4 gene of complex I in the respiratory chain results in interference with the transfer of electrons by complex I and concomitantly acts to pump protons from the stroma up into the inter-membrane cavity. Thus, in 7S fibroblasts, glycolysis predominates over mitochondrial ATP production, and results in compensatory changes in cell size to include more mitochondria, albeit with damage and poor function (fig. 8D). Relative to PDL 15 (black solid line), the diameter of 7S fibroblasts was about 1.5 times larger than NHDF to PDL 25, and the cell size increase was doubled and the final size increased to about 3 to 8 times to PDL 35 (fig. 8D, left panel).
Consistent with the functional recovery of 7SP cells after mtDNA replacement, the significant increase in cell size observed in early PDL was inhibited in 7SP fibroblasts after mitochondrial replacement (fig. 8D, right panel). In addition, the size of 7SP cells that received mitochondrial replacement of exogenous mitochondria in PDL 8 was maintained up to the 50 th PDL (fig. 8D, right panel). In addition, by the 10 th PDL, the concentration of Citrate Synthase (CS) was 2 times higher than the CS concentration in NHDF cells in 7SP fibroblasts, consistent with the increase in size increase of 7SP fibroblasts (data not shown).
To confirm that the observed improvement in cell function following mitochondrial replacement was not due to contamination by other cell types, a Short Tandem Repeat (STR) assay was performed that could identify cells from different sources (fig. 8E). Importantly, at different time points, STR types in mitochondria-replaced cells were identical to those of original 7SP fibroblasts (fig. 8E), indicating no contamination. In addition, RT-PCR showed that transfer of exogenous mitochondria derived from cells expressing telomerase and E6 did not convert primary fibroblasts into cancer cells (fig. 8F).
Overall, these results indicate that transfer of exogenously isolated mitochondria with wild-type mtDNA to mitochondria in 7SP cells derived from Leigh syndrome patients increases the lifespan of 7SP cells and improves cellular function. Importantly, the metastasis does not convert the mitochondria-replaced 7SP cells into cancer cells.
Example X: transfer of outer mitochondria to fibroblasts from Leigh syndrome patients results in functional mitochondria
The functional role of mitochondrial replacement in 7SP fibroblasts was further evaluated by analyzing the respiratory function of the cells by using orobos O2k (fig. 9A). The results quantitatively indicate that after mitochondrial replacement of cell production, basal respiration and ATP production (free daily activity) continued to decrease from PDL 10 to PDL 20, and the maximum capacity of the electron transport system maintained the original level of 7SP fibroblasts (fig. 9B). By PDL 30 after exogenous mitochondrial transfer, all 3 indices of respiratory function (conventional, ETS and free daily activity) were elevated and even exceeded the primitive cellular level (fig. 9B). These results indicate that there is a short delay in reconstituting the electron delivery system with healthy and non-mutated complex I after mtDNA replacement. Proton leakage showed the same kinetics as non-mitochondrial ATP production, which improved from early stabilization (fig. 9B).
These results demonstrate that transfer of exogenous mitochondria to fibroblasts from patients with mitochondrial diseases or disorders can produce functional mitochondria.
Example XI: transfer of exogenous mitochondria can dissipate long-term, sustained Reactive Oxygen Species (ROS) production
To identify the nature of 7SP fibroblast-derived MirC, both reperfusion and starvation models under culture conditions were used for 7SP fibroblast-derived MirC, original 7SP fibroblasts and NHDF as control. These stress conditions cause apoptosis in cultured cells, the extent of which can be quantified by annexin v (annexin v) as an early marker and Propidium Iodide (PI) as a late marker. Among the environmental hazards, reperfusion injury is primarily due to mitochondrial dysfunction. Cells susceptible to mitochondrial dysfunction due to mtDNA mutations are more vulnerable to reperfusion injury than healthy cells.
The cells were cultured at 1X 105Each cell was seeded in 6-well plates. On day 2, 600. mu. M H were added2O2(FUJIFILM Wako Pure Chemical) was added to the cells for reperfusion model or serum-free DMEM (FUJIFILM Wako Pure Chemical) without essential amino acids ("-EAA") was used as the culture medium for starvation model. At 3h H 2O2Or hunger for 48hAfter starvation, the cells were washed with PBS and collected into centrifuge tubes. Annexin V-FITC and PI solutions were added to the cells and allowed to react for 30 min at room temperature in the absence of light. Cells were then rapidly FCM analyzed using 488 and 561nm laser lines. Fluorescence data were collected using SH800 (Sony). Flow cytometry files were analyzed by using FlowJo software (TreeStar).
The results indicate that 7SP cells derived from Leigh syndrome subjects are stressed in two ways (i.e., H)2O2And starvation) are highly sensitive. As shown in FIGS. 10A-10D, using H2O2Treated 7SP cells showed a significant increase in early and late apoptosis. In the reperfusion model (H)2O2) NHDF did not show any significant damage during apoptosis based on annexin v (annexin v) and PI staining (fig. 10B-fig. 10D). However, this mild reperfusion stress caused apoptosis in 7SP fibroblasts. In contrast, the positive fraction of both annexin v (annexin v) and PI in 7SP fibroblast-derived MirC was significantly lower than parental 7SP fibroblasts and close to the level of NHDF cells (fig. 10B-fig. 10D). Importantly, there was no significant difference between 7SP fibroblast-derived MirC and NHDF, suggesting that MirC regained the ability to tolerate this mild reperfusion injury.
The same trend as reperfusion was identified using the starvation model (fig. 10E-10H). In 7SP fibroblasts, high apoptosis was shown in both early and late stages, while 7SP fibroblast-derived MirC showed almost the same level of apoptosis as NHDF (basal values), which was significantly smaller than those in the original 7SP fibroblasts (fig. 10F-fig. 10H). These results further confirm that the mitochondrial replacement method of the present invention improves functional recovery of recipient cells.
These results demonstrate that transfer of exogenous mitochondria from healthy cells to cells with mutant mtDNA can improve the function of recipient cells.
Example XII: transfer of exogenous mitochondria to recipient cells restores early senescence-associated secretory phenotype (SASP)
This example demonstrates that transfer of exogenous mitochondria to recipient cells restores the early senescence-associated secretory phenotype (SASP). SASPs, which are composed of inflammatory cytokines, growth factors, and proteases, are characteristic of senescent cells.
To determine whether the transfer of exogenous mitochondria to senescent cells could restore SASP, the expression levels of representative SASP cytokines IL-6 and IL-8, chemokine CXCL-1, and growth factor ICAM1 were quantitatively measured at the transcript level for NHDF, 7SP fibroblasts, and 7SP fibroblast-derived MirC cells (which had almost the same PDL, from about 15 to 20) (fig. 11). In 7SP fibroblasts, IL-6 was significantly higher than those in NHDF and 7SP fibroblast-derived MirC, whereas the other 3 factors did not show any significant difference in these cells. In this PDL, 7SP fibroblasts did not show typical SASP, but only IL-6 expression was higher, indicating early senescence. Importantly, in this PDL by 7SP fibroblast-derived MirC, the early senescence process can be restored.
Overall, these data demonstrate that mitochondrial replacement not only can treat mitochondrial diseases with mtDNA mutations, but can also rejuvenate senescent cells, such as cells involved in a range of diseases including neurodegenerative, cardiovascular, metabolic, and autoimmune diseases, and even cancer.
Example XIII: iPS cells generated from mtDNA-substituted fibroblasts
To determine whether induced pluripotent stem cells (ipscs) can be generated using cells derived from patients with long deletions of mtDNA, we attempted to construct ipscs from 7SP fibroblasts using standard methods with noct 3/4, Klf4, Sox2 and c-myc (oksm) sendai virus suitable for NHDF. A protocol design chart is provided in fig. 12A.
Detection of the iPSC colonies at early stages alkaline phosphatase staining (AP staining) showed that the original 7SP fibroblast-derived colonies appeared to have a disrupted appearance at day 21, whereas the mitochondrially replaced 7SP fibroblast-derived colonies were robust (figure 12B). In contrast, not receiving mtDNA substitutionFibroblasts do not produce any aggregatesAnd (fig. 12B). Some iPS cell lines were able to be generated from mitochondrially replaced 7S fibroblasts as measured by AP staining (fig. 12C and 12D). iPSC clones were stable in culture and showed similar morphology between individual colonies (figure 12E). Immunohistochemical staining confirmed the expression of human pluripotent stem cell markers SOX2, OCT3/4, NANOG, SSEA4, TRA1-81 and TRA1-60 on mitochondria-replaced 7SP fibroblast-derived colonies overexpressing OKSM (fig. 12F).
Ipscs generated by the methods described herein were further compared to commercially available KYOU-DXR0109B human Induced Pluripotent Stem (IPS) cells [201B7 ]. Importantly, mitochondria-replaced 7SP fibroblasts showed the same effective level as iPS production by healthy fibroblasts. In addition, in agreement with previous studies, qPCR of 12S-rRNA normalized for nuclear β -actin showed that iPS cells produced by mitochondria-replaced 7SP fibroblasts showed half the mtDNA content relative to control and mtDNA levels were similar to those of the 201B7 iPSC standard (fig. 12G).
Furthermore, the level of hmt10158 heterogeneity in the ipscs generated was less than 10% (fig. 12H). Quantification of absolute mtDNA copy number confirmed a decrease in mtDNA levels and a decrease in mutant mtDNA (fig. 12I).
These results demonstrate that ipscs can be generated using the mitochondrial replacement techniques provided herein, and that they can be adapted for use in the clinical field, as the complete procedure uses only clinically appropriate materials.
Example XIV: mitochondrial replacement of mitochondria from donor cells alters the lifespan of recipient cells
This example demonstrates that mtDNA replacement can alter the lifespan of recipient cells. To validate the hypothesis that mitochondrial replacement can rejuvenate senescent cells, two models were evaluated with respect to cell cycle capacity, such as doubling time and PDL at growth arrest.
Models were designed using NHDF and TIG1 embryonic lung cells with early PDL (about 5 to 10, referred to as "young") and late PDL (about 40 to 45, referred to as "old"). One model included young cells replaced with mitochondria from old cells, denoted as "O2Y", and the other model included old cells replaced with mitochondria from young cells, denoted as "Y2O" (fig. 13A).
The extent of mtDNA crossover was assessed by TaqMan SNP genotyping assay based on the single nucleotide difference of mtDNA at position 16145 between NHDF and TIG1 (which are a and G, respectively). NHDF-derived MirC clearly showed that more than 90% of the endogenous mtDNA (hmt16145-A) was replaced with TIG 1-derived mtDNA (hmt16145-G) (FIG. 13C). A small percentage of hmt16145-a detected in parental TIG1 cells was considered a background error (fig. 13C).
In addition, the Y2O model clearly showed a restoration of lifespan in old cells to approximately 65PDL (fig. 13D). Control old cells and empty transfectants showed growth arrest at 55PDL, consistent with the Hayfick limit. On the other hand, O2Y showed a reduction in the lifespan of young cells to about 45PDL (fig. 13E). The difference of about 10PDL in the two models can be attributed to exogenous mtDNA. These results demonstrate that the transfer of exogenous mitochondria from young cells to old cells can rejuvenate the cells.
Example XV: optimisation of mitochondrion-replaced cells from human primary T cells (MirC) transfected with mRNA
This example describes the generation of mitochondrially replaced cells (MirC) from human primary T cells transfected with mRNA.
Prior to the experiments, the use of human primary T cells was approved by our ethical committee. Peripheral blood was drawn from healthy volunteers and centrifuged at 400g, 20 degrees for 35 minutes using percoll of specific gravity 1.077 to separate lymphocytes. Will be 1 × 106Individual cells/ml of isolated lymphocytes were plated onto 96-well plates coated with anti-CD 3 and anti-CD 28 antibodies. Plates were prepared by incubating 5. mu.g/ml anti-CD 3 and 1. mu.g/ml anti-CD 28 overnight and pre-heated at 37 ℃ 2 hours prior to inoculation. One day after inoculation, IL-7 and IL-15 were added to the medium at concentrations of 20. mu.g/ml and 10. mu.g/ml, respectively. The medium was replaced every 3 days with the same concentrations of IL-7 and IL-15 as the initial addition.
Transfection was performed using a MaxCyte electrotransformation machine, which complies with GMP/GCP standards, according to the manufacturer's protocol. mRNA was generated in the mMESSAGE mMACHINE T7 Ultra kit (Thermo Fisher) according to the manufacturer's protocol and with minor modifications. Briefly, after endonuclease digestion, a DNA template of mRNA was prepared from a plasmid having a DNA sequence without refining the fragment to reduce the possibility of rnase mixing as low as possible (fig. 14A).
The results indicate that the unrefined DNA template for mRNA production of EGFP resulted in near 100% gene transfer efficiency with high expression and high viability 24 hours after gene transfection (fig. 14B and 14C). Antibiotic selection is not required using this method due to high transfection efficiency. In addition, transfection of MTS-XbaIR resulted in a decrease in mitochondrial membrane potential, which could be attributed to a decrease in endogenous mtDNA (FIG. 14D).
To determine the optimal protocol with respect to the timing of isolated mitochondrial co-incubation, fluorescence images of human primary T cells receiving GFP mRNA by electroporation using MaxCyte ATX were collected over a period of 8-days, as shown (fig. 14E). Fluorescence images of control electroporated cells in which cells were transfected with GFP plasmid (fig. 14F, top panel) show similar kinetics as in fibroblasts. Expression peaked on day 2 and disappeared by day 8. In contrast, cells receiving MTS-GFP mRNA (fig. 14F, lower panel) showed higher expression within 4 hours after electroporation and disappeared earlier on day 6 than those in cells transferred with the plasmid.
Protein expression of GFP in cells receiving MTS-GFP mRNA was assessed by immunoblot analysis using capillary electrophoresis. Peak expression occurred at day 4, and expression disappeared at day 6, as shown in the immunoblot (fig. 14H) and quantified in fig. 14G. Kinetic quantification of XbaIR transcript levels was performed by qPCR and showed that endonuclease transcript expression was highest 4 hours after gene transfer (fig. 14I). XbaIR transcript levels decreased rapidly by day 2 and were negligible by day 6 (fig. 14I). Mitochondrial content was estimated by quantifying 12S rRNA (fig. 14J) and showed that mitochondria decreased to about 30% by day 2 and remained at less than 20% throughout the experiment.
Overall, these results indicate that mRNA transfection with an endonuclease fused to MTS, such as XbaI, can efficiently degrade host mtDNA and can be used to generate mitochondrially replaced cells (MirC) from human primary T cells.
Example XVI: production of mitochondrially replaced cells from human primary T cells (MirC) transfected with mRNA
After determining the best point in time for mitochondrial transfer in human primary T cells from example XV, mitochondrial co-incubation was performed on day 7 to prevent digestion of exogenous mtDNA by any remaining endonuclease. Figure 15A shows the MirC protocol for human primary T cells.
To determine the heterogeneity of mtDNA in recipient human primary T cells after mitochondrial replacement, mtDNA differences between donor mitochondria and recipient cells were determined by TaqMan SNP genotyping assay. Sequencing of the D-loop of mtDNA in normal human primary T cells and EPC100 (mitochondrial donor cells) showed a difference in 2 nucleotide positions (nucleotides 218 and 224mtDNA), which are C/C and T/T for T cells and EPC100 cells, respectively (fig. 15B). To trace the standard curve for the TaqMan SNP genotyping assay, a variable region fragment encompassing nucleotides 218 and 224 of mtDNA was subcloned into pBluescript SK (-). Polymorphic nucleotides were located on the human cambridge reference sequence and primers and probes were designed to amplify and target the desired region of the D-loop, where the probes had FAM and VIC fluorophores (fig. 15C). qPCR was performed using TaqMan polymerase with 5' exonuclease activity and the threshold cycle number (Ct value) was determined and fitted to a standard curve generated using several different copy numbers of the above plasmids for each sequence. After somatic mitochondrial replacement, EPC100 mtDNA sources were predominant in human T cells on days 7 and 12, while free-running chromosomes receiving electroporation but no genetic material and co-incubated with isolated mitochondria in the same protocol as MirC showed less than 10% exogenous source of mtDNA on day 7 and background levels on day 12 (fig. 15D). This indicates that MTS-XbaIR mRNA is favorable for efficient mitochondrial transfer in human primary T cells.
Then, in order to evaluate the effect of mitochondrial transfer on MirC human T cell function, a respirometry experiment was performed using orobos O2 k. The results showed that ATP production and coupling efficiency recovered in human T cell-derived MirC at day 7, whereas ρ (-) human T cells generated by transfer using electroporated XbaIR mRNA maintained the loss of ATP production throughout the experiment (fig. 15E). Representative raw data using the coupling-control protocol (CCP) are shown in fig. 15F and 15G, and it is shown that MirC T cells are able to restore mitochondrial respiration.
These results demonstrate that human primary T cells are capable of mitochondrial replacement to produce MirC using a GMP-grade electrotransformation instrument, such as the one produced by MaxCyte inc.
Example XVII: production of mitochondrially replaced cells from mouse primary T cells (MirC) transfected with mRNA
Further characterization of T cell-derived MirC was performed on murine T cells. Isolation of murine T CELLs from suspension solutions from the spleen was performed using the EasySep mouse isolation kit (STEM CELL Technologies, Inc.), which provided a highly purified T CELL population by negative selection using a magnet. Isolated murine T cells (1X 10) 6Individual cells/ml) were seeded onto 96-well plates with Dynabeads mouse T-activator CD3/CD28(Invitrogen, Inc.) and recombinant IL-2 at a bead-to-cell ratio of 1: 1. The media used to culture murine T cells was determined relative to cell growth and CD3 expression and RPMI1640 was found to be superior to TexMACS (fig. 16A). For example, viability and total cell number were higher in cells cultured in RPMI1640 compared to TexMACS. The medium was changed every 3 or 4 days.
Electroporation of murine T cells was then performed using a Nucleofector instrument and mRNA. The kinetics of GFP expression after mRNA transfer was similar to that of human T cells (fig. 16B). For example, almost all cells were found to strongly express GFP 6 hours after electroporation of MTS-GFP mRNA (FIG. 16B). This demonstrates that MTS-GFP is efficiently transfected. GFP intensity decreased rapidly with time and eventually disappeared on day 6 after electroporation (fig. 16B).
Transfection of MTS-XbaI mRNA showed that murine T cells showed less expression of XbaIR transcripts relative to human T cells and continued even at low levels up to day 6 (fig. 16C). Quantification of 12S rRNA levels as surrogate markers for mtDNA indicated that murine mtDNA remained at approximately 40% of controls even on day 6 (fig. 16D). Then, co-incubation of Ds-red labeled exogenous mitochondria and rho (-) murine T cells was performed on day 5 (FIG. 16E). FACS analysis of engulfed fluorescence-labeled mitochondria revealed a positive fraction (9.73%) of T cells expressing the outer source mitochondria 48 hours after co-incubation with isolated mitochondria, despite longer duration of XbaIR and lower level of endogenous mtDNA reduction relative to human T cells (fig. 16F). The percentage of positive cells expressing exogenous mitochondria was even higher than in fibroblast experiments, indicating that this procedure for murine T cells may be optimal to produce T cell-derived mircs.
Example XVIII: transfer of exogenous mitochondria to T cells reverses senescence
This example demonstrates that mitochondrial replacement is successful in murine T cells and rejuvenated senescent T cells.
To assess whether exogenous mitochondria could be successfully transferred to produce murine T cell-derived MirC, the level of mtDNA heterogeneity was of BL6 (recipient) cells and NZB (donor) cells. Two consecutive polymorphisms of 2766 and 2767mtDNA were verified for ND1 (fig. 17A). Specifically, BL6 mitochondria contained AT positions 2766 and 2767 of mtDNA, whereas NZB mitochondria contained GC AT the same position. The primer set and two probes were designed to distinguish polymorphisms using different fluorophores for each of the GC and AT polymorphisms (fig. 17B). In addition, two different plasmids were generated to express the GC and AT polymorphisms, respectively, and a standard curve was generated to aid in the quantitative determination of heterogeneity in MirC.
Quantification of mitochondrial replacement (XbaIR Mt) in BL6 cells transfected with MTS-XbaI and co-incubated with mitochondria isolated from NZB mice demonstrated that exogenous mtDNA was predominant, whereas free-standing chromosomes co-incubated with isolated mitochondria did not engulf any exogenous mtDNA in the absence of endonuclease mRNA after electroporation (fig. 17C). This result demonstrates that T cells are permissive for mitochondrial replacement.
Since the results described herein demonstrate that fibroblast-derived mircs can undergo in vitro rejuvenation (fig. 13), T cell-derived mircs were also examined for rejuvenation potential. Recipient cells from murine T cells were prepared from the spleen of mice greater than 80 weeks old (C57BL/6), and donor murine mitochondria were isolated from the liver of mice about 10 weeks old (C57 BL/6). Telomere length has been reported to decrease with age. Therefore, telomere length was measured by using absolute mouse telomere length quantitative qPCR assay kit (ScienCell, Inc.). After treatment of murine cells with MTS-XbaIR mRNA and co-incubation with exogenous mitochondria from young donor cells to produce MirC (young to old: YtoO), a 1.7-fold increase in telomere length relative to naive old T cells was observed (FIG. 17D). This indicates that the mitochondria replaced cells showed rejuvenation characteristics.
In addition, SASP was evaluated using the same representative marker panel described previously (fig. 11). Measurement of CXCL1, ICAM1, IL-6 and IL-8 showed that murine T cell-derived MirC reduced IL-6 and CXCL-1 and showed no change in ICAM-1 and IL-8 (FIG. 17E). These results indicate that for MirC T cells, SASP is reduced.
Furthermore, it has been found that senescent T cells exhibit a higher DNA Damage Response (DDR) compared to young T cells. Thus, DDR was measured using histone 2A (H2A) phosphorylation antibody for MirC and naive T cells. The results showed that the positive fraction of DDR was lower in MirC (1.53%) compared to naive T cells (4.75%) (fig. 17F). Thus, MirC T cells have lower DDR levels, indicating reversal of senescence behavior.
These in vitro results support that somatic mitochondrial substitutions were validated in MirC mtDNA, and that the substitutions resulted in a variety of alterations that indicate senescence reversal in MirC T cells.
Example XIX: tumor growth reduction by Adoptive Cell Transplantation (ACT) using old T cell-derived MirC containing exogenous mitochondria from young mice
To examine the functional potential of mitochondrial replacement to rejuvenate senescent cells, Adoptive Cell Transplantation (ACT) experiments were performed. The AE17 mesothelioma cell line derived from the peritoneal cavity of C57BL/6J mice injected with asbestos fibers was used to develop tumors in the mice. Previous experiments using this model have shown that tumor growth is reduced by ACT from young syngeneic T cells, rather than by ACT from old syngeneic T cells (Jackaman et al OncoImmunology 2019; 8(4): 1-16).
To determine whether rejuvenated old T cells produced by transferring isolated exogenous mitochondria from young mice to T cells from old mice showed functional activity, AE17 cells were injected subcutaneously into 3 groups of old mice (group 1: old mice with ACT from T cells from young mice; group 2: old mice; or group 3: old mice with ACT from MirC of T cells derived from old mice transferred exogenous mitochondria from young mice) (fig. 18A). The ability of old T cell-derived MirC to inhibit tumor growth was evaluated. 22 to 24 month old C57BL/6 mice were used in ACT experiments. Young mice used in the experiment were 2 to 3 month old mice. In addition to body weight measurements, tumor growth was measured using NIH images of photographs taken every 3 days (fig. 18B). On day 14, 2X 10 suspended in 100. mu.L Matrigel 6AE17 inoculation of individual cells and T cell transfer is currently considered day 0. On day 0, 2X 106Young or old T cell-derived mircs of individual cells were injected intravenously into tumor-bearing mice. On the same day, one intraperitoneal injection of recombinant IL-2 (2. mu.g) was performed, followed by two more injections on days 2 and 3.
Body weight in each group showed no significant difference (fig. 18C). However, tumors were reduced in group 1 mice (old mice with young T cells) and group 3 mice (old mice with old T cell-derived mircs), while tumors were stably grown in group 2 (empty) mice (fig. 18D). The relative mean masses showed a similar trend to individual mice, indicating that MirC behaved similarly to young T cells (fig. 18E).
To verify the presence of infused T cells in the animals, T cells derived from GFP transgenic mice were implanted into syngeneic C57BL/6 mice, and peripheral blood and spleen were examined to follow the donor cells (fig. 18F). Two-dimensional plots with FSC versus FL-1 were generated to illustrate the rare population for detection of GFP fluorescence. Negative controls (top left) using C57BL/6 mice and positive controls (bottom left) using GFP transgenic mice were generated for both peripheral blood and spleen (FIG. 18G). A defined population of T cells expressing GFP fluorescence was identified in both samples, although the fractions were 0.057% and 0.9% in peripheral blood and spleen, respectively (fig. 18G). In this protocol, metastatic T cells were detected on day 6 post-transplantation (fig. 18H), which verifies that this protocol can be used to evaluate the capacity of metastatic cells. In addition, the percent chimerism after exogenous T cell infusion was found to increase when higher numbers of cells were infused (fig. 18I).
These results clearly demonstrate that ex vivo MirC production using mitochondria from young mice into T cells from old mice can act effectively in vivo and reduce tumor burden at similar levels as T cells from young mice.
Example XX: hematopoietic stem cells capable of producing MirC
To date, methods of gene transfer of hematopoietic stem cells have primarily involved the use of viral vectors, as the target is primarily a genetic disorder requiring sustained gene expression of defective genes. Therefore, electroporation is not used in current protocols for gene transfer of hematopoietic stem cells due to the need to produce permanent gene expression. In contrast, the mitochondrial replacement techniques provided herein are aimed at achieving transiently high expression of exonucleases.
Based on experiments with fibroblasts and T cells, nuclear transfection (Nucleofector)/electroporation using mRNA was adjusted for conditions and several conditions were examined for murine fetal liver-derived Sca-1 positive cells, which are considered to be an enriched population of Hematopoietic Stem Cells (HSCs), fig. 19A. Of several conditions, three conditions (codes from machine suppliers: programs X-001, Y-001 and T-030) were evaluated by immunofluorescence and cell viability (FIG. 19A). Depending on the procedure used (procedures X-001, Y-001 and T-030, respectively), the experimental conditions were designated MTS-GFP1, 2 and 3.
On day 1 after electroporation of mRNA with GFP, the Mean Fluorescence Intensity (MFI) was further analyzed by FACS analysis (fig. 19B). The results indicate that the optimal conditions are the X-001 program (MTS-GFP1) because although the MFI shifts to the right little in this condition, it is more pronounced than the others (FIG. 19B). Murine bone marrow-derived Sca-1 cells were co-cultured with mitochondria isolated from syngeneic murine cells, a stable gene-modified cell line expressing DsRed fluorescence. 3-D fluorescence images of bone marrow-derived Sca-1 cells 48 hours after co-incubation showed that the exogenous mitochondria were engulfed (FIG. 19C). Mitochondrial transfer efficiency was estimated for DsRed fluorescence axis by FACS analysis and showed that about 10% of the Sca-1 subpopulation showed a right shift in fluorescence, indicating that BM-derived Sca-1 positive cells can undergo somatic mitochondrial replacement (fig. 19D). However, the transfer of exogenous mitochondria in MTS-GFP expressing cells without endogenous mitochondrial ablation is too low for clinical use.
Then, we examined whether this mitochondrial replacement procedure by generation of ρ (-) cells transferred using MTS-XbaIR mRNA can be applied to hematopoietic stem cells (FIG. 19E). Authentic hematopoietic stem cell populations are considered c-kit +、Sca-1+、Lineage-、CD34-(referred to as KSLC) which account for about 0.005% of total bone marrow cells (Wilkinson, A.C. et al Nature,571(7763):117-121 (2019)). After FACS sorting of KSL cells from murine bone marrow-derived cells (fig. 19F), KSL cells were cultured for 5 days in the presence of stem cell factor and TPO, as well as polyvinyl alcohol (PVA). Macroscopically, KSL cells maintained morphology and showed a short doubling time of 19 hours (fig. 19G).
As described above, heterogeneity changes were evaluated using TaqMan SNP genotyping assay. The assay protocol is shown in fig. 19H. Murine KSLC-derived MirC demonstrated 99.9% of exogenous mtDNA with polymorphism in NZB at day 6 after endonuclease mRNA transfer by electroporation (FIG. 19I), which indicates that the exogenous mtDNA almost completely replaced the endogenous mtDNA of CL57 BL/6. These results demonstrate that hematopoietic stem cells allow this technology to produce MirC.
Example XXII: micro-droplet digital PCR (ddPCR) for mtDNA and heterogeneity measurements
This example demonstrates that the presence of specific mtDNA sequences, such as mutations in mtDNA, can be determined for mitochondrial dna (mtDNA) using digital pcr (dpcr). Micro-droplet digital PCR (ddpcr) is a method for performing digital PCR based on water-oil emulsion droplet technology.
Primary dermal fibroblasts from patients with mitochondrial disease were analyzed. Table 1 below provides patient information.
Table 1: patient information
Cells from the target population are encapsulated into droplets at a concentration of 1 cell per droplet, along with a PCR mixture comprising primers and probes. Cell density was optimized to produce single cells in a single droplet, and fibroblasts were grown at 1 × 106Individual cells/mL of final dilution were used for ddPCR. After single cell encapsulation, cell lysis and amplification of the target sequence are performed within the droplets. The number of droplets with a fluorescent signal indicates the number of cells with the target or reference gene.
Briefly, 20 × primer/probe mixtures were prepared as described in table 2 below. The standard ddPCR master mix (master mix) is a 25. mu.L mix containing the above primer/probe mix, template DNA and 2 XddPCR super mix (super mix).
Table 2: dPCR primer and probe mixture
20 Xprimer/Probe mixtures
Volume per 100. mu.L (μ L)
100 mu M F1 primer
10
100 mu M R1 primer
10
100 μ M labeled probe
5
PCR grade water
75
Table 3: dPCR reaction premix
Reagent
Volume of reaction per 25. mu.L (. mu.L)
2 XDddPCR super mix
12.5
20 Xprimer/Probe mixtures
1.25
Stencil (100 ng/. mu.L)
1
PCR grade water
10.25
Samples were loaded into 8-chamber cartridges (cartridges) using 20 μ L of prepared qPCR samples followed by 70 μ L droplets in adjacent wells to produce oil. A rubber pad is stretched across the top of the chamber to ensure a vacuum seal. Each 8-chamber cartridge was loaded onto a QX100 droplet generator, producing 20,000 droplets per sample. Using a 50 μ Ι multichannel pipettor, 40 μ Ι _ of the resulting droplets were transferred to a 96-well plate and heat sealed with a pierceable foil. The plate was placed in a thermal cycler at a 50% (3 ℃/sec) ramp rate using standard 2-step qPCR thermal cycling conditions. Prior to performing thermocycling conditions, the primer/probe set was optimized using a temperature gradient to optimize the annealing/extension temperature.
Table 4: dPCR cycling conditions
After thermal cycling, the plates were loaded onto a QX100 droplet plate reader and the end point reactions were analyzed. Statistical analysis of poisson for the number of positive and negative drops yielded absolute quantification of the target sequence.
Before examining diseased cells, the specificity of probes designed for mutated sequences and the sensitivity of probes designed for non-mutated sequences were evaluated by using normal human dermal fibroblasts (NHDF cells) with non-mutated sequences (identical to the cambridge reference sequence) (fig. 20A-fig. 20C). The dots in the lower left area indicate no cells in the droplet. Evaluation of the 3 different sets of probe sets clearly detected non-mutated sequences (bottom right in BK01 (fig. 20A), top left in BK02 (fig. 20B), and top left in BK04 (fig. 20C)), and no mutated sequences (top left in BK01 (fig. 20A), BK02 (fig. 20B), and bottom right in BK04 (fig. 20C)).
ddPCR of fibroblasts from BK01 indicated a small percentage of double positive populations, and most were homoheterogeneous cells with mutant mtDNA (fig. 20D). There is no apparent population of homoeogeny with non-mutated mtDNA in single cells. In addition, BK02 showed a small fraction of double positive cells, indicating heterogeneity at the single cell level, defined as micro-heterogeneity (fig. 20E). The results of BK02 show a major population of homoeogeny of mutant mtDNA and no population with homoeogeny of non-mutant mtDNA was identified.
Taken together, these results indicate that homotypic heterogeneity and heterogeneity can be accurately and quantitatively evaluated at the single cell level. In addition, the results demonstrate that mtDNA can be accurately measured in subjects with mitochondrial disorders, which can be used to evaluate therapeutic compositions prior to subject transplantation or to monitor mtDNA content before and/or after therapy.
Example XVII: mtDNA replacement of bone marrow-derived mesenchymal stromal cells (BM-MSCs) from donor cGMP production in recipient hematopoietic stem or progenitor cells (HSPCs).
This example demonstrates that hematopoietic stem or progenitor cells (HSPCs) can be modulated ex vivo using mtDNA replacement methods for therapy provided herein.
HSPC regulation may be performed ex vivo in conjunction with stem cell transplantation. Briefly, peripheral blood stem cells are mobilized and a blood sample is obtained from a patient. Isolation of peripheral hematopoietic stem or progenitor cells (HSPC), e.g., CD34+The cells are sent to a production facility. At the production facility, mitochondria are partially eliminated according to the methods provided herein.
Donor mitochondria were isolated using bone marrow-derived mesenchymal stromal cells (BM-MSCs) produced by modern good manufacturing criteria (cGMP) from cell banks (e.g., Waisman Biomanufacturing). Initial bone marrow aspirates were collected under full informed consent and according to federal regulations (e.g., 21 CFR 1271). Aspirates were processed under cGMP conditions and binned early in generations for subsequent amplification.
Donor mitochondria from BM-MSCs are transferred to cultured HSPCs, altering heterogeneity. The modified HSPCs were returned to the medical centre for autologous transplantation (i.e. to the same subject that sequestered the HSPCs). Prior to transplantation, the patient receives minimal treatment, which may include a non-myeloablative regimen, such as topical irradiation or sub-lethal doses of anti-cancer drugs, such as busulfan. The modified HSPCs containing only allogeneic donor mitochondria are returned to the patient.
This example demonstrates that HSPC can be modulated ex vivo using mtDNA replacement methods for therapies provided herein that do not include transplantation of allogeneic HSPC.
The embodiments described above are intended to be exemplary only, and those skilled in the art will recognize, or be able to ascertain using no more than routine experimentation, numerous equivalents to the specific compounds, materials, and procedures. All such equivalents are considered to be within the scope of the invention and are encompassed by the following claims.
Sequence listing
<110> Emel BIOTHERAPEUTICS (IMEL BIOTHERAPEUTICS, INC.)
<120> METHODS AND COMPOSITIONS FOR TREATING MITOCHONDRIAL DISEASEs OR DISORDERS AND heterogeneity (METHODS AND COMPOSITIONS FOR TREATING MITOCHONDRIAL DISEASE OR disorder AND heterogeneity)
<130> 14595-001-228
<140>
<141>
<150> 62/817,987
<151> 2019-03-13
<150> 62/731,731
<151> 2018-09-14
<150> 62/718,891
<151> 2018-08-14
<160> 47
<170> PatentIn version 3.5
<210> 1
<211> 24
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic oligonucleotides
<400> 1
tcatccatgg ggacgagaag ggat 24
<210> 2
<211> 24
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic oligonucleotides
<220>
<221> modified base (modified _ base)
<222> (15)..(15)
<223> a, c, t, g, unknown or otherwise (a, c, t, g, unknown or other)
<400> 2
tcatccatgg gggcnagaag ggat 24
<210> 3
<211> 24
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic oligonucleotides
<400> 3
tcatccatgg ggacgagaag ggat 24
<210> 4
<211> 24
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic oligonucleotides
<400> 4
tcatccatgg gggcgagaag ggat 24
<210> 5
<211> 15
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic probes
<400> 5
cttctcgtcc ccatg 15
<210> 6
<211> 25
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic primers
<400> 6
agccatttac cgtacatagc acatt 25
<210> 7
<211> 16
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic probes
<400> 7
cccttctcgc ccccat 16
<210> 8
<211> 200
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic polynucleotides
<400> 8
cagtacatag tacataaagc catttaccgt acatagcaca ttacagtcaa atcccttctc 60
gtccccatgg atgacccccc tcagataggg gtcccttgac caccatcctc cgtgaaatca 120
atatcccgca caagagtgct actctcctcg ctccgggccc ataacacttg ggggtagcta 180
aagtgaactg tatccgacat 200
<210> 9
<211> 20
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic primers
<400> 9
cacggaggat ggtggtcaag 20
<210> 10
<211> 53
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic oligonucleotides
<400> 10
accacaactc aacggctaca tagaaaaatc caccccttac gagtgcggct tcg 53
<210> 11
<211> 53
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic oligonucleotides
<400> 11
accacaactc aacggctaca tagaaaaacc caccccttac gagtgcggct tcg 53
<210> 12
<211> 24
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic oligonucleotides
<400> 12
acatagaaaa acccacccct tacg 24
<210> 13
<211> 24
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic oligonucleotides
<400> 13
acatagaaaa acccacccct tacg 24
<210> 14
<211> 24
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic oligonucleotides
<400> 14
acatagaaaa atccacccct tacg 24
<210> 15
<211> 24
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic oligonucleotides
<400> 15
acatagaaaa atccacccct tacg 24
<210> 16
<211> 200
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic polynucleotides
<400> 16
tataaatagt accgttaact tccaattaac tagttttgac aacattcaaa aaagagtaat 60
aaacttcgcc ttaattttaa taatcaacac cctcctagcc ttactactaa taattattac 120
attttgacta ccacaactca acggctacat agaaaaatcc accccttacg agtgcggctt 180
cgaccctata tcccccgccc 200
<210> 17
<211> 26
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic primers
<400> 17
caacaccctc ctagccttac tactaa 26
<210> 18
<211> 18
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic probes
<400> 18
acatagaaaa atccaccc 18
<210> 19
<211> 18
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic probes
<400> 19
ctacatagaa aaatccac 18
<210> 20
<211> 18
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic primers
<400> 20
gtcgaagccg cactcgta 18
<210> 21
<211> 12
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic oligonucleotides
<400> 21
tacttaacca cc 12
<210> 22
<211> 12
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic oligonucleotides
<400> 22
tacttgacca cc 12
<210> 23
<211> 40
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic oligonucleotides
<400> 23
caagcaagta cagcaaccaa ccctcaacta tcacacatca 40
<210> 24
<211> 40
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic oligonucleotides
<400> 24
caagcaagta cagcaatcaa ccttcaacta tcacacatca 40
<210> 25
<211> 400
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic polynucleotides
<400> 25
attctaattt aaactattct ctgttctttc atggggaagc agatttgggt accacccaag 60
tattgactca cccatcaaca accgctatgt atttcgtaca ttactgccag ccaccatgaa 120
tattgtacgg taccataaat acttgaccac ctgtagtaca taaaaaccca atccacatca 180
aaaccccctc cccatgctta caagcaagta cagcaatcaa ccctcaacta tcacacatca 240
actgcaactc caaagccacc cctcacccac taggatacca acaaacctac ccacccttaa 300
cagtacatag tacataaagc catttaccgt acatagcaca ttacagtcaa atcccttctc 360
gtccccatgg atgacccccc tcagataggg gtcccttgac 400
<210> 26
<211> 22
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic oligonucleotides
<400> 26
ctctgttctt tcatggggaa gc 22
<210> 27
<211> 21
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic oligonucleotides
<400> 27
atccatgggg acgagaaggg a 21
<210> 28
<211> 23
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic oligonucleotides
<400> 28
ccttgttccc agaggttcaa atc 23
<210> 29
<211> 24
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic oligonucleotides
<400> 29
tctttattaa tatcctaaca ctcc 24
<210> 30
<211> 29
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic oligonucleotides
<400> 30
aaaggatccc cttgttccca gaggttcaa 29
<210> 31
<211> 25
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic oligonucleotides
<400> 31
tgttctttat taatgcccta acact 25
<210> 32
<211> 200
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic polynucleotides
<400> 32
gcgtaagact taaaaccttg ttcccagagg ttcaaatcct ctccctaata gtgttcttta 60
ttaatatcct aacactcctc gtccccattc taatcgccat agccttccta acattagtag 120
aacgcaaaat cttagggtac atacaactac gaaaaggccc taacattgtt ggtccatacg 180
gcattttaca accatttgca 200
<210> 33
<211> 20
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic oligonucleotides
<400> 33
tgccgtatgg accaacaatg 20
<210> 34
<211> 41
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic oligonucleotides
<400> 34
gaatggggac gaggagtgtt aggatattaa taaagaacac t 41
<210> 35
<211> 41
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic oligonucleotides
<400> 35
gaatggggac gaggagtgtt agggcattaa taaagaacac t 41
<210> 36
<211> 28
<212> PRT
<213> Unknown
<220>
<223> description unknown: COX10 MTS peptide
<400> 36
Met Ala Ala Ser Pro His Thr Leu Ser Ser Arg Leu Leu Thr Gly Cys
1 5 10 15
Val Gly Gly Ser Val Trp Tyr Leu Glu Arg Arg Thr
20 25
<210> 37
<211> 24
<212> PRT
<213> Unknown
<220>
<223> description unknown COX8 MTS peptide
<400> 37
Met Ser Val Leu Thr Pro Leu Leu Leu Arg Ser Leu Thr Gly Ser Ala
1 5 10 15
Arg Arg Leu Met Val Pro Arg Ala
20
<210> 38
<211> 17
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic probes
<400> 38
agcaatcaac cttcaac 17
<210> 39
<211> 15
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic probes
<400> 39
caaccaaccc tcaac 15
<210> 40
<211> 22
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic primers
<400> 40
ccccatgctt acaagcaagt ac 22
<210> 41
<211> 22
<212> DNA
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic primers
<400> 41
ttggagttgc agttgatgtg tg 22
<210> 42
<400> 42
000
<210> 43
<400> 43
000
<210> 44
<400> 44
000
<210> 45
<211> 17
<212> PRT
<213> Artificial sequence
<220>
<223> description of artificial sequences: synthetic peptides
<400> 45
Met Asn Ser Thr Val Asn Phe Gln Leu Thr Ser Phe Asp Asn Ile Gln
1 5 10 15
Lys
<210> 46
<211> 47
<212> PRT
<213> Artificial sequence
<220>
<223> description of Artificial sequences synthetic polypeptides
<400> 46
Met Asn Phe Ala Leu Ile Leu Met Ile Asn Thr Leu Leu Ala Leu Leu
1 5 10 15
Leu Met Ile Ile Thr Phe Trp Leu Pro Gln Leu Asn Gly Tyr Met Glu
20 25 30
Lys Ser Thr Pro Tyr Glu Cys Gly Phe Asp Pro Met Ser Pro Ala
35 40 45
<210> 47
<211> 50
<212> PRT
<213> Artificial sequence
<220>
<223> description of Artificial sequences synthetic polypeptides
<400> 47
Met Phe Phe Ile Asn Ile Leu Thr Leu Leu Val Pro Ile Leu Ile Ala
1 5 10 15
Met Ala Phe Leu Thr Leu Val Glu Arg Lys Ile Leu Gly Tyr Met Gln
20 25 30
Leu Arg Lys Gly Pro Asn Ile Val Gly Pro Tyr Gly Ile Leu Gln Pro
35 40 45
Phe Ala
50